CSF1R inhibition for the depletion of tumor-associated macrophages (TAMs) seemed like a promising therapeutic strategy, but while TAM depletion occurred as expected, antitumor efficacy has been incredibly limited. To understand why, Kumar et al., in a study published in Cancer Cell, revealed the additional immunosuppressive mechanism activated by CSF1R inhibition and exploited this information to create an effective anti-tumor strategy.
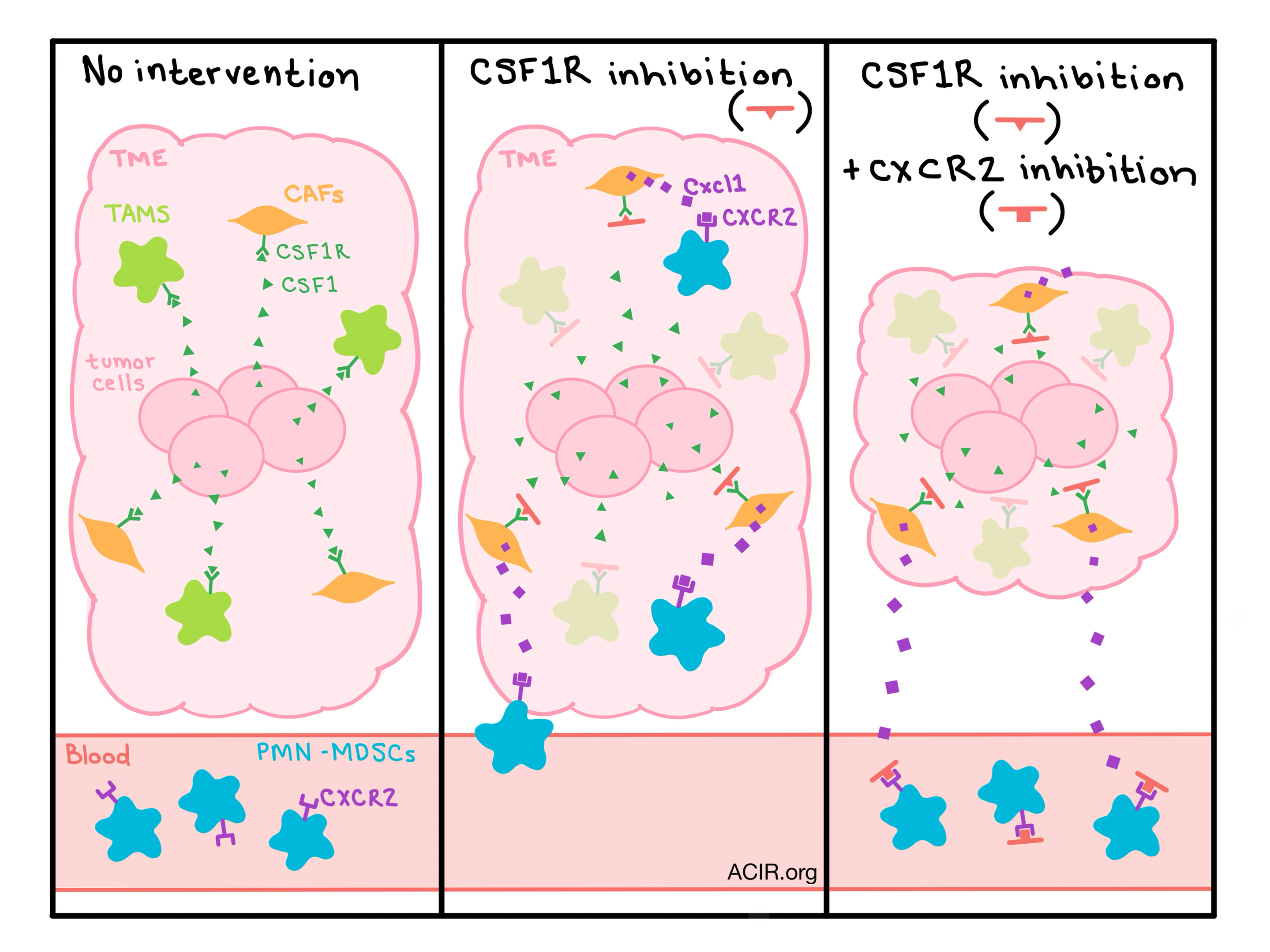
One way that tumors support immunosuppressive TAMs is by producing CSF1, which is recognized by TAMs via the receptor CSF1R. This pathway supports differentiation and survival of TAMs, so researchers hypothesized that inhibiting CSF1R would deplete TAMs and delay tumor growth. In preclinical models, various small molecule inhibitors have been tested, and while they appear to successfully deplete TAMs, little to no anti-tumor effects have been observed. Kumar et al. tested the CSF1R inhibitor JNJ-40346527 on a variety of tumor models; in addition to depletion of TAMs, the researchers also observed accumulation of granulocytes with potent immunosuppressive activity, which were identified as polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs). Further, the team was able to prove that the increase in PMN-MDSCs was due to enhanced recruitment to the tumor site, rather than proliferation of precursor cells.
To understand how this second pro-tumor immune cell type was being recruited to the tumor, Kumar et al. evaluated chemokines in tumor cell lysates following CSF1R inhibition and noticed an upregulation of chemokines, most notably Cxcl1, that are known to recruit granulocytes via the CXCR2 receptor. Carcinoma-associated fibroblasts (CAFs) were identified as the major source of Cxcl1 and the researchers unexpectedly found that CAFs also express the CSF1R protein on their cell surface and in the cytoplasm. A series of mechanistic experiments showed that CSF1 activates the CSF1/CSF1R pathway on CAFs, causing the histone deacetylase HDAC2 to migrate to the Cxcl1 promoter and silence the production of Cxcl1. Blocking this pathway with a CSF1R inhibitor reinstates Cxcl1 expression and increases the recruitment of tumorigenic PMN-MDSCs to the tumor site, abrogating any effect of TAM depletion.
In humans, CXCL1 is not as potent a recruiting cytokine as Cxcl1 in mice, however, evaluation of human tumors following surgical resection showed that CAFs produced high levels of CXCL8, which is known for recruiting neutrophils. The researchers went on to show that in addition to recruiting neutrophils, CXCL8 also plays a role in recruiting PMN-MDSCs. Gene silencing and neutralizing antibody experiments demonstrated that CXCL8 production is controlled by the CSF1/CSF1R pathway.
In an effort to prevent the recruitment of tumor-promoting PMN-MDSCs following CSF1R inhibition, Kumar et al. tested a CSF1R inhibitor in combination with a CXCR2 inhibitor. Neither single agent showed antitumor efficacy, but together they resulted in a highly significant delay in tumor progression in two mouse models. Evaluation of the tumors showed that the combination successfully decreased the presence of TAMs and abrogated the CSF1R inhibitor-induced accumulation PMN-MDSCs. The combination therapy also enhanced the therapeutic effect of anti-PD-1 therapy, resulting in a dramatic antitumor effect.
Speculating on why tumors would contribute to a mechanism that inherently prevents the recruitment of pro-tumor PMN-MDSCs, Kumar et al. suggest that early in tumor development, infiltration of highly inflammatory neutrophil populations might be damaging to the nascent tumor; thus, early-stage tumors might utilize CSF1 production to limit this population until the tumor is more advanced.
Kumar et al. show that the use of CSF1R inhibitors to deplete immunosuppressive TAMs and slow the growth of tumors is not a lost cause as long as the recruitment of PMN-MDSCs to the tumor site is addressed. Further, they show that this therapy may enhance the efficacy of anti-PD-1 therapy, which could one day lead to improved clinical efficacy.
by Lauren Hitchings



