
Last week, the ACIR team attended the AACR Tumor Immunology and Immunotherapy conference in Miami, Florida. This week’s extensive special feature covers select talks from the conference.
Topics include:
Keynote Speakers
Tumor and Host Factors
T Cell Analysis
Chimeric Antigen Receptors (CARs)
Understanding Exhaustion
Activating the Immune Response
The Tumor Immunology and Immunotherapy conference, hosted by AACR in Miami, Florida, began with talks from two keynote speakers. Elizabeth Jaffee opened the conference by observing that tumors with high mutation burden or DNA repair deficiency are typically more responsive to immunotherapy, and that novel strategies are desperately needed to better target tumors without these advantageous characteristics. Jaffee discussed some of the strategies that are gaining steam, including targeting the STING pathway, utilizing OX40 agonists, vaccinating against neoepitopes, mindfully combining the checkpoint blockades (choosing the right doses, timing, and order), and modifying treatment plans based on evolving tumor and immune information for more precise tumor control.
Next, Carl June provided an overview of progress in CAR T cell engineering and addressed current research that is tackling some of the remaining challenges, particularly attacking solid tumors. Revealing some data from his own ongoing research, June introduced the possibility of using CAR T cells that express unshielded 7SL RNA, an inflammatory stimulus, to enhance immune cell infiltration and antitumor efficacy in solid tumors. Interestingly, June also discussed testing emerging immunotherapies in pet dogs diagnosed with cancer, as both a way to save canine lives and to study immunotherapy in a setting that more closely resembles humans.
In a talk on tumor and host factors that regulate antitumor immunity, Thomas Gajewski provided an overview of immune inflamed and immune excluded tumors and discussed how systemic immunity and tumor-intrinsic factors may shape tumor features, clinical responses, and immune escape. Exploring how the microbiome shapes antitumor immunity, Gajewski cited a series of experiments in which mice from different suppliers showed differential tumor growth; co-housing the mice, performing fecal transplant, or administering a mix of bifidobacterium bacteria, however, neutralized that effect. Based on a strong correlation between response and a particular microbial sequence signature in a small cohort of melanoma patients treated with anti-PD-1, Gajewski showed that human fecal transplants from responding patients, but not non-responding patients, improved antitumor immunity and outcomes in germ-free mice, providing evidence that strategies to manipulate the microbiome (dietary changes, probiotics/antibiotics, fecal transplants) might improve (or inhibit) antitumor immunity. Gajewski did note that not all differences in the susceptibility of strains could be attributed to differences in the microbiota, however, and so he closed his talk by providing genetic crossing evidence that mice from the supplier Harlan contain a single recessive susceptibility gene, not yet identified.
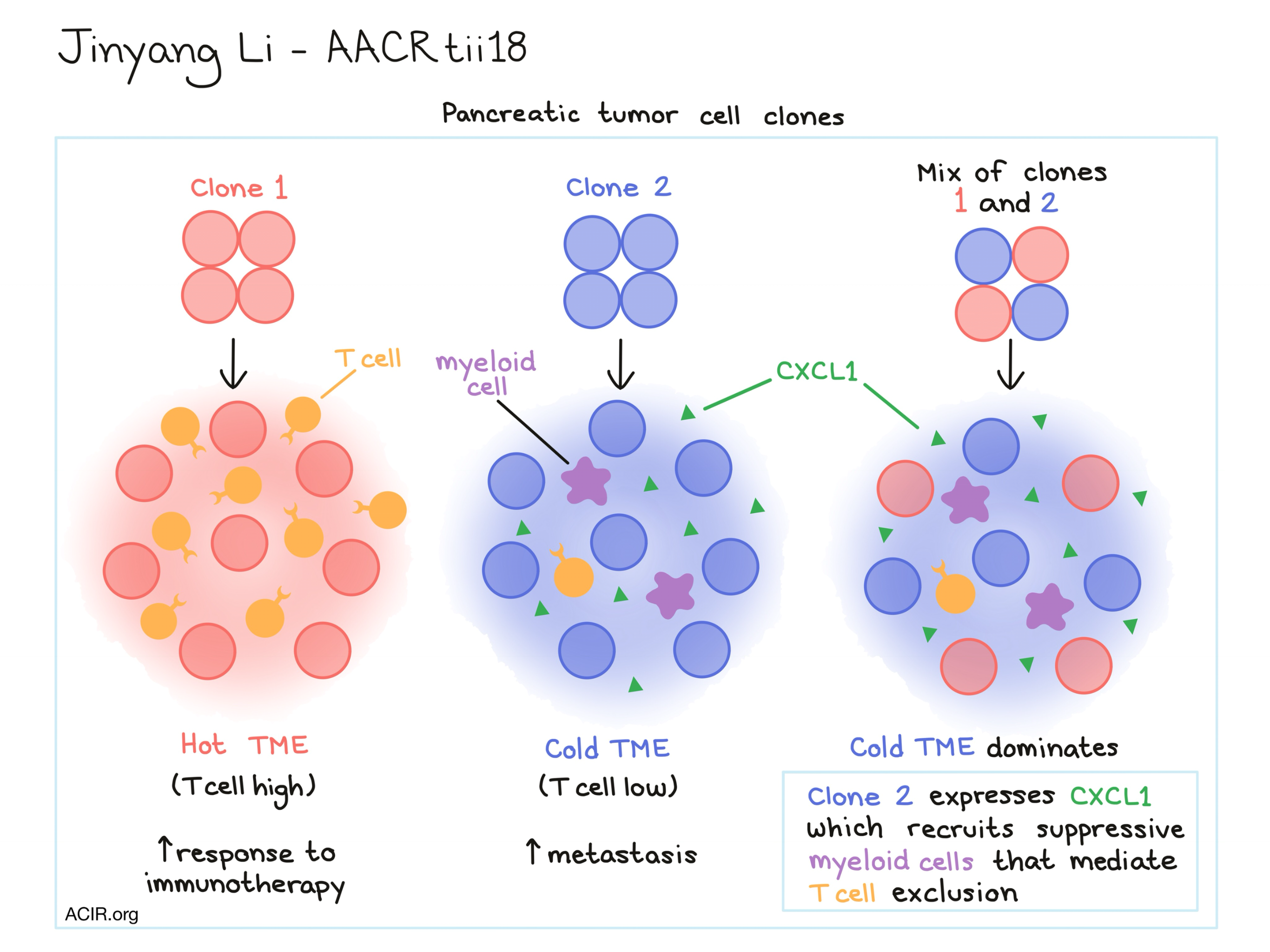
Discussing how tumor cell-intrinsic factors underlie heterogeneity in the immune infiltrate, Jinyang Li showed that pancreatic tumors cell lines exhibit heterogeneous TIL levels and that certain tumor cell clones can maintain a consistent level of immune infiltration. Li and his team were able to consistently generate tumors that would have either low or high T cell infiltration. Tumors with low T cell infiltration showed increased metastatic activity, while tumors with high T cell infiltration were more sensitive to immunotherapy. When Li and his team generated a tumor by mixing the TIL-high and TIL-low tumor cell types, the TIL-low tumor type dominated and the mixed tumor became cold. As it turned out, the TIL-low tumor cell clones were marked by increased expression of CXCL1 which was responsible for the recruitment of suppressive myeloid cells, which were in turn responsible for mediating T cell exclusion.
Lisa Coussens discussed the role of chronic inflammation and the complement cascade in antitumor immunity. She showed that characteristics of inflammation are tissue-specific and that chronic inflammation is established early. Using mesothelioma and pancreatic adenocarcinoma as examples, she showed how the complexity of inflammation is tumor- and tissue-specific, reflecting a balancing of TH2 and TH1 status. Depending on the tumor/tissue, either CD4+ T cells or B cells were involved, affecting resident monocyte/macrophage populations which ultimately impacted effector or resident memory T cells. Coussens identified tissue-specific patterns in myeloid responses and showed that licensing of antitumor immune cells was driven by humoral immunity. Coussens also discussed the role of the redundant complement cascade as an alternative path to activating pro-tumor macrophages or mast cells, based on the HPV oncogene model system in which progressive inflammation led to spontaneous tumors. She also identified the activation of complement factor C5aR1 as a pro-tumor immune program and showed that inhibition of C5AR1 could reverse resistance to chemotherapy through the activation of TH1-polarizing macrophages to increase effector memory and resident memory T cells in the tumor bed.
Paul Thomas reported that the use of the algorithm TCRdist to compare TCRs and study motifs that confer cross-reactivity revealed that TCRs recognizing epitopes with just a single amino acid difference between them are not actually very similar. This suggests that TCRs recognizing self antigens would be unlikely to identify neoepitopes and therefore central tolerance to native peptides may not limit the ability of the TCR repertoire to respond to mutated peptides. To understand what else might limit antitumor T cell responses, Thomas and his team analyzed tumors and T cells from from pediatric ALL patients with very low mutation burdens to compare this to the very low rate (2%) of responses to predicted neoepitopes found in high mutation burden tumors. Surprisingly, all ALL patients were found to have endogenous CD8+ T cells that recognized tumor neoantigens, and a high fraction (70-80%) of the predicted neoepitopes were detected among patient CD8+ T cells. They also observed immunodominance hierarchies among neoepitope-specific CD8+ T cells, which may be a more significant factor in high mutation burden tumors and contribute to the low rate of observation. Together this research suggests that even patients with low tumor mutation burdens may benefit from neoantigen-targeting immunotherapy. Further, the team identified responses to a potential public neoepitope derived from the ETV6-Runx1 fusion protein (which arises from a driver mutation), indicating the potential for a more generalized TCR T cell-based therapy for ALL.
In an effort to make better sense of tumor immune complexity and heterogeneity, Ido Yofe discussed his analysis of RNA and TCR sequencing data from nearly 30,000 tumor-infiltrating T and NK cells. Yofe and his team created an unbiased map, which identified two subsets of effector T cells – only one of which becomes exhausted or “dysfunctional”. They observed an anti-correlation between cytotoxicity and dysfunctionality and could not correlate the dysfunctionality with other tumor or immune parameters. Looking at T cell clonality, they found that T cell expansion and dysfunction were interrelated and that dysfunctional cells were clonally distinct from effectors, and were tumor reactive and highly proliferative, indicating that they might still be playing a part in the immune response.
Chimeric Antigen Receptors (CARs)
CAR T cells are effective against liquid tumors, but have thus far been ineffective against solid tumors due to limited trafficking and suppressive factors in the tumor microenvironment (TME). Michael Klichinsky discussed his group’s unique use of CARs to transform macrophages rather than T cells. Based on the abundance of macrophages in solid tumors and their capacity for antitumor effector functions and antigen presentation, Klichinsky hypothesized that M1-polarized (antitumor) CAR macrophages would target and phagocytose tumor cells, and then process and present tumor antigens to elicit a T cell response. In support of the first arm of his hypothesis, Klichinsky showed video evidence of CAR macrophages effectively phagocytosing targeted tumor cells – sometimes more than one at a time. To test for in vivo efficacy, Klichinsky administered HER2-targeting CAR macrophages to mice bearing HER2+ ovarian cancer. A single dose of CAR macrophages induced antitumor efficacy and an improved survival in about half of mice. Klichinsky also showed CAR macrophages upregulated costimulatory ligands and antigen-presenting genes, leading to enhanced T cell stimulation. Interestingly, CAR macrophages were also resistant to conversion to M2 (pro-tumor) phenotype by immunosuppressive cytokines, further supporting their potential to maintain efficacy in a solid TME. Based on this promising data, a Phase I clinical trial of HER2-directed CAR macrophages has been planned.
As part of a normal immune response, T cells responding to a threat become exhausted and less functional. To find out exactly how T cells become exhausted, and what exactly “exhausted” means, John Wherry and his team studied T cells recovered following acute and chronic infection on the epigenetic level to better understand the transcriptional reprogramming process. Interestingly, principal component analysis of open chromatin regions showed that exhausted cells were transcriptionally quite distinct from both naive and effector/memory cell clusters, indicating that exhaustion arises as a separate pathway. Following checkpoint blockade, exhausted T cells could be temporarily reinvigorated via re-wiring of transcriptional networks, but they could not be reprogrammed into memory cells. Digging into the mechanism of exhaustion induction, comparative gene expression analysis identified the transcription factor Tox as the “pioneer” for exhaustion. Tox was found to open enhancers upstream of additional transcription factors, including TCF1. NFAT signaling appeared to induce Tox expression in T cells, which then became self-sustained. Downregulation of Tox prevented the formation of exhausted cells without disrupting T cell activation or the formation of memory/effector cells, indicating that Tox could potentially be used as a therapeutic target.
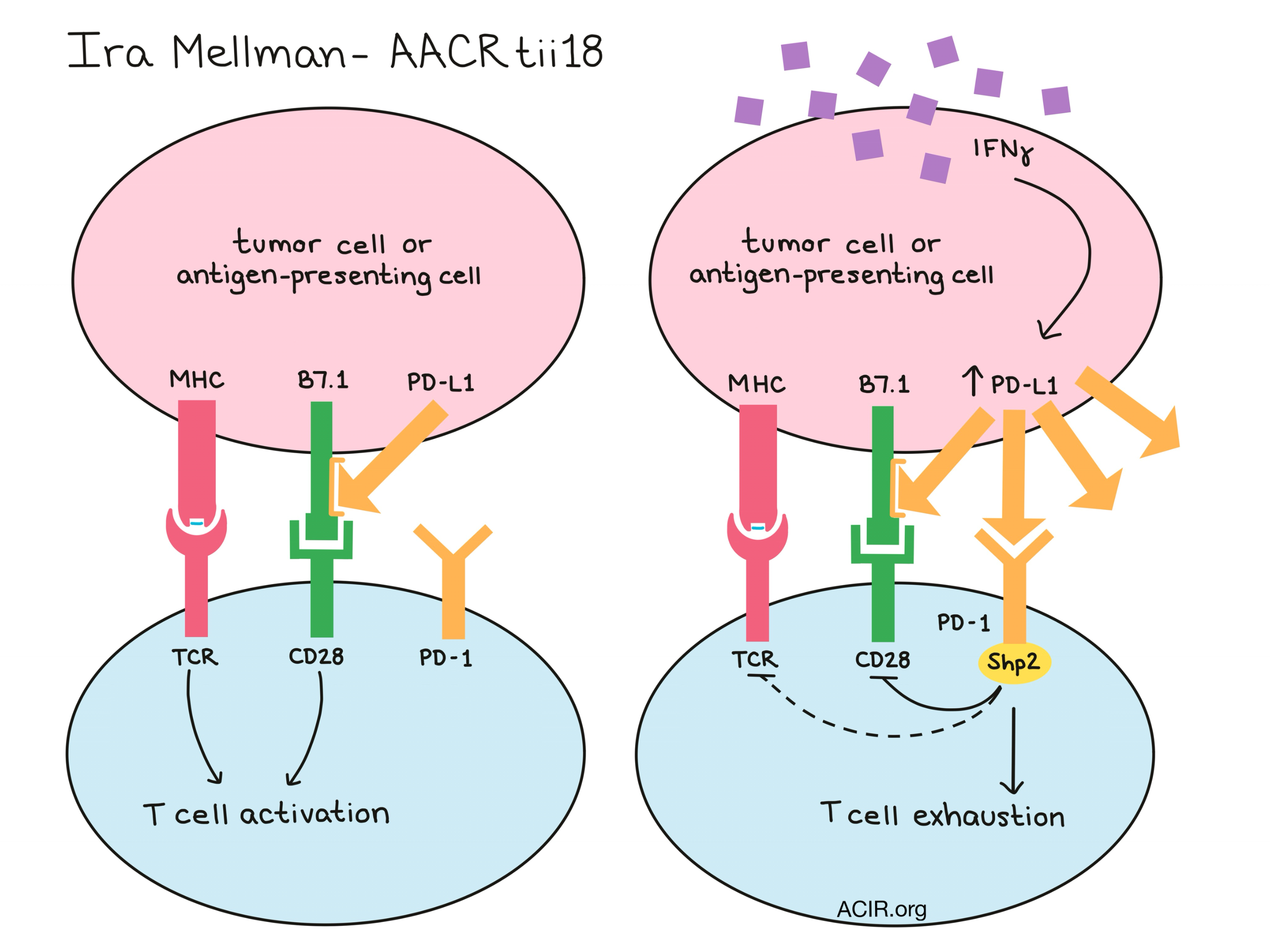
Also challenging the current paradigm of T cell exhaustion, Ira Mellman discussed research on the mechanism by which PD-1 signaling induces hallmarks of exhaustion. Mellman showed that PD-1 signaling inhibits CD28 from within the T cell via sequestration of the Shp2 phosphatase, which would ultimately dephosphorylate CD3ζ and CD28. Mellman also showed that the efficacy of anti-PD-L1 in CT26 tumors was dependent on the CD28/B7 pathway. B7.1 is a key ligand for binding costimulatory CD28, but Mellman found that it also binds in cis to PD-L1 on tumor and myeloid cells; this interaction prevented PD-L1 from interacting with with PD-1 on T cells, however, it did not prevent B7.1 from interacting with CD28. In cases of increased IFNγ signaling where PD-L1 becomes upregulated, the PD-1/PD-L1 interaction would become dominant and T cells would become exhausted. Conditional PD-L1 knockout experiments also revealed a dominant role for CLEC9A+ dendritic cells in control of PD-L1- tumors, which led to the hypothesis that PD-1/PD-L1 inhibition may be predominantly working through expansion of memory and/or effector T cells, rather than through the reversal of exhaustion in existing cells. This was supported by TCR sequence analysis after PD-L1 blockade, which showed clonal expansion of memory and effector T cell populations in both tumor and adjacent normal tissues, and a strong positive correlation between clonal expansion and survival.
Activating the Immune Response
Weiwen Deng discussed the use of ADU-S100, a novel cyclic dinucleotide (CDN) analog that activates the STING pathway, to initiate tumor-mediated T cell priming in situ. As an intratumorally administered monotherapy, ADU-S100 elicited robust, local, tumor-specific CD8+ T cell responses at both low and high doses. The low (immunogenic) dose resulted in limited tumor control, and the high (ablative) dose eliminated local tumors, but led to systemic drug distribution and compromised systemic immunity. In mice bearing dual-flanked 4T1 or MC38 tumors, injecting one tumor with a single immunogenic dose of ADU-S100 enhanced systemic immunity and sensitized previously unresponsive mice to PD-1 blockade, leading to eradication of both injected and non-injected tumors and near-complete responses, dependent on CD8+ T cells. In the poorly immunogenic B16F10 tumor model, the addition of ADU-S100 to combination anti-PD-1 and anti-CTLA-4 induced tumor-specific CD8+ T cell responses and improved tumor control, yielding several complete and durable responses. Cured mice were protected from rechallenge. ADU-S100 is currently being tested in clinical trials in combination with checkpoint blockade.
In an effort to generate an immune response using a neoantigen-targeted vaccine, Karin Lee harvested tumors from two mice bearing MC38 colon carcinoma and sequenced them for non-synonymous mutations, which varied between the two tumors. Lee identified 51 neoepitopes that were shared between tumors, and from those, selected top affinity neoepitopes and synthesized them for vaccination. Administration of a four-epitope neoantigen-targeted vaccine did not produce a survival benefit, however, combining the vaccine with the IL-15 superagonist N-803 and anti-PD-L1 increased the immunogenicity of the vaccine and showed initial antitumor efficacy. The addition of NHS-IL-12 (a tumor necrosis-targeted version of IL-12) increased CD8+ T cell infiltration, improved activation and effector profiles, allowed for long-term maintenance of immunogenicity, and induced tumor regression. In testing of different vaccine formulations, a multi-epitope adenovirus vaccine (encoding all four epitopes in one construct) outperformed admixed adenovirus vaccines (encoding individual neoepitopes separately). Only the multi-epitope vaccine led to epitope spread to other MC38 epitopes.
Overall, the AACR Tumor Immunology and Immunotherapy conference served as a great meeting of minds for researchers from across immune-oncology. The field of immunotherapy is in full bloom, and sharing what’s new and what’s next is one of the best ways to move forward and make progress in the fight against cancer.
by Lauren Hitchings



