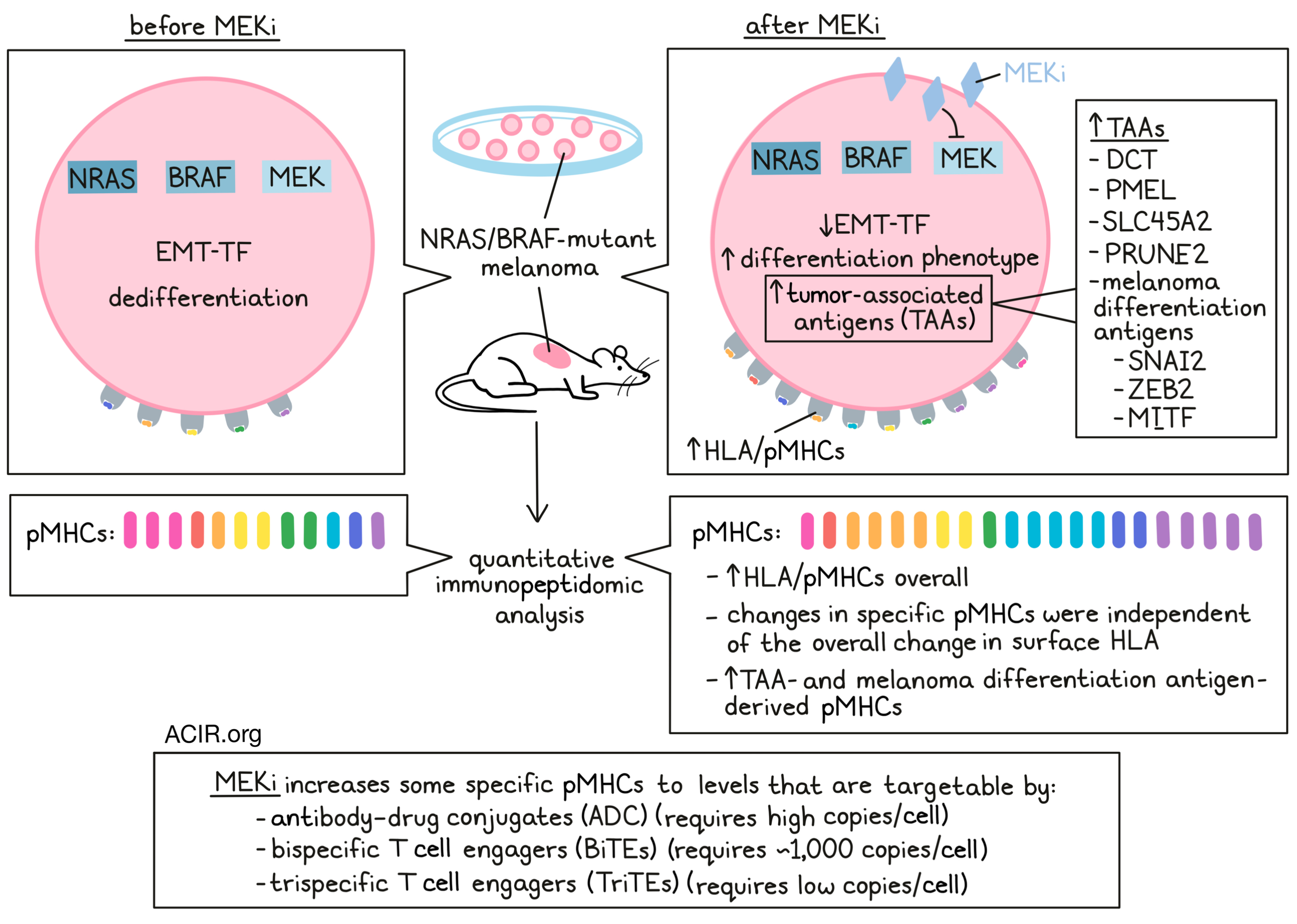
In NRAS/BRAF-mutant melanoma, MEK and BRAF inhibitors (MEKi/BRAFi) are known to increase expression of tumor antigens and upregulate pMHCs, leading to increased antitumor immune responses. However, MEK/BRAF inhibitors alone are not usually effective long-term, and when used in combination with immunotherapies, can cause high toxicity. To better understand the effects of MEKi/BRAFi and explore the potential to use them in safer and more effective ways, Stopfer and Rettko et al. performed quantitative immunopeptidomic analysis to measure relative changes in pMHC repertoires following MEKi in vitro and in vivo. Based on their findings, they evaluated several drug combination strategies. Their results were recently published in PNAS.
To begin, the researchers selected two NRAS- and four BRAF-mutant cell lines with a range of sensitivities to the MEKi, binimetinib. Following treatment with binimetinib, quantitative immunopeptidomic profiling showed a median increase in pMHC levels across HLA-A, -B, and -C. In contrast, mass spectrometry showed a wide distribution in presentation levels across peptides, with some peptides showing strong upregulation, and others showing strong downregulation, independent of the overall change in surface HLA.
Investigating which peptides were selectively enriched relative to the median change in pMHC presentation, Stopfer and Rettko et al. found that two enriched peptides in the SKMEL5 line were derived from the well known tumor-associated antigens (TAAs) DCT and PMEL. Upon further investigation, this observation reflected a general pattern of dose-dependent increased expression of TAA-derived peptides following MEKi. This was consistent across cell lines and in the context of a different MEKi, trametinib (another MEKi), suggesting a mechanistic basis for this effect.
Next evaluating whether this pattern held true in vivo, the researchers evaluated the effects of MEKi on four different melanoma cell lines in mice. Quantitative multiplexed immunopeptidomics analysis of pMHCs isolated from tumor cells at early time points after treatment showed that mean HLA presentation was altered. In BRAF-mutant lines, combination of MEKi with the BRAFi, encorafenib, showed similar or increased changes in pMHC compared to MEKi alone. TAA analysis showed TAA enrichment in at least one treatment condition across models, with melanoma differentiation antigens showing notable enrichment above median fold changes as well. This high abundance of TAAs mapped to peptides correlated with the highest changes in gene expression, further validating the differential expression of TAA-derived pMHCs.
The researchers found that MEKi-induced pMHC presentation could not be predicted using overall transcript or protein expression datasets alone, suggesting that post-translational processing may play a key role in determining peptide presentation. Despite the lack of a general correlation, clustering analysis of changes in pMHC, protein, and RNA expression revealed a subset of source genes, including the melanoma differentiation antigens, that showed a clear connection between pMHC presentation and changes in transcription, translation, and protein degradation.
Typically, MAPK activation induces EMT transcription factors that promote de-differentiation (low MITF) and tumorigenesis, while inhibition of this pathway can reverse the EMT-TF phenotype back to a differentiation phenotype (high MITF). In line with this, gene-set enrichment analysis showed that MEKi treatment reduced enrichment of the epithelial-to-mesenchymal transition (EMT) pathway. The researchers also observed increased differentiation and decreased tumorigenic marker transcript expression following MEK inhibition. This was further reflected in pMHCs, with increased pMHCs derived from differentiation-associated proteins like SNAI2, ZEB2, and MITF following MEKi. TCGA data also showed evidence of these relationships, with MITFlow tumors showing lower HLA expression, and patients experiencing reduced survival. MITF also correlated with melanoma differentiation antigen expression, suggesting that MITFlow NRAS/BRAF-mutant tumors may benefit from MEKi or other MAPK inhibitors to induce a MITFhi differentiation phenotype with enhanced TAA pMHC expression.
Next, the researchers hypothesized that MEKi could enhance responses to immunotherapies targeted against MEKi-modulated antigens. To explore this, they performed absolute quantification experiments to estimate copies per cell for 18 HLA-A*02:01 epitope targets that increased in presentation following MEKi in SKMEL5 cells. All 18 peptides were quantifiable in MEKi-treated SKMEL5 cells, with 14 and 11 quantifiable in A375 and RPMI-7951 cells. respectively. Copies-per-cell estimations varied widely, even for epitopes derived from the same source proteins and presented on the same HLA alleles – observations that would not have been evident based on bulk analysis. Based on this data, the researchers selected four HLA-A*02:01-associated TAAs (derived from SLC45A2, PMEL, DCT, and PRUNE2) with high MEKi-induced abundance in SKMEL5 cells as targets for antibody generation. SLC45A2, PMEL, DCT were chosen for further analysis based on suitable biophysical properties and high differential expression in melanoma compared to healthy skin cells.
An antibody–drug conjugate (ADC) consisting of anti-SLC45A2 and MMAF (a tubulin polymerization inhibitor) showed enhanced efficacy against MEKi-treated SKMEL5 cells compared to control-treated cells. However, no difference was observed in RPMI-7951 cells, as they likely did not express high enough pMHC levels, even after treatment with MEKi. To target less abundant pMHCs, the researchers then generated bispecific T cell engagers (BiTEs) by fusing antibody Fabs to an anti-CD3 scFv. Treatment with each of the 3 BiTEs showed that BiTE-mediated T cell activation was generally enhanced following MEKi due to higher target expression. Even RPMI-7951 cells, which were unresponsive to the SLC45A2-ADC, yielded a strong cytotoxic response and increased cell death in cells treated with MEKi and the SLC45A2-BiTE. In some cell lines with high target pMHC expression at baseline, BiTEs induced similar responses regardless of MEKi pretreatment. Overall, it seemed that BiTEs were most effective when pMHC targets were present at levels of at least 1000 copies/cell – at least an order of magnitude lower than the threshold for ADC activity. BiTEs could also be combined to generate trispecific T cell engagers (TriTEs) to enhance cytotoxic responses at even lower concentration thresholds, allowing for effective targeting of some pMHCs even without MEKi pretreatment.
Together these results suggest that employing quantitative immunopeptidomics can reveal changes in peptide presentation and provide new insights compared to bulk analysis. Using these tools revealed that MEKi can be used to boost TAA presentation and enhance antitumor immune responses through restoration of a MITFhigh differentiation phenotype in tumor cells. The enhanced presentation of TAA-derived peptides could then be specifically exploited using antibody–drug conjugates, bispecific/trispecific T cell engagers, and potentially other targeted immunotherapies to improve tumor control in NRAS/BRAF-mutant tumors.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, co-first author Lauren E. Stopfer answered our questions.

What was the most surprising finding of this study for you?
We often think of pMHC targets at a protein level (ex. gp100/PMEL) and not an epitope level (peptide1 versus peptide2). Here we show that while there is an expected large range of copy-number densities across different tumor antigens, even antigens derived from the same source protein exhibit a wide range of surface densities (ex. A*02:01 PMEL epitopes vary by 100x in measured copies per cell). Furthermore, the amount that presentation levels change with drug treatment is also highly variable (PMEL epitopes increase 2-6x in presentation with MEK inhibition). This highlights that not all antigens (even from the same protein) are created equal!
What is the outlook?
We hope that this work and the methods described help facilitate new studies to explore the impact of different therapies on the antigen landscape across disease types, and that the specific findings here – largely that MAP kinase inhibitors boost presentation of select melanoma tumor antigens – help to rationally guide how best to combine small molecules inhibitors and immunotherapies.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently traveled to Barcelona to speak at a proteomics symposium and had the chance to visit a vineyard in Catalonia to learn about the history of Cava (sparkling wine) and how it is made. In honor of my trip, when this paper was published, I toasted with a glass of Cava instead of Champagne!



