Aiming to understand the mechanism behind CD4+ T cell help in regulating CD8+ T cell differentiation in the context of chronic infection and cancer, Zander and Schauder et al. utilized single-cell RNA sequencing (scRNAseq) to analyze the transcriptional profiles and differentiation pathways of CD8+ T cells during chronic viral infection. The results, recently published in Immunity, indicate that CD4+ T cells support the formation of a highly cytotoxic subset of CD8+ T cells via production of IL-21.
The researchers began by performing scRNAseq analysis on CD8+ T cells that were specific for the GP33-41 peptide of the lymphocytic choriomeningitis virus (LCMV) at 8 and 30 days post infection with the Cl13 chronic strain of LCMV. The analysis identified nine clusters, including the following three major clusters observed at Day 30:
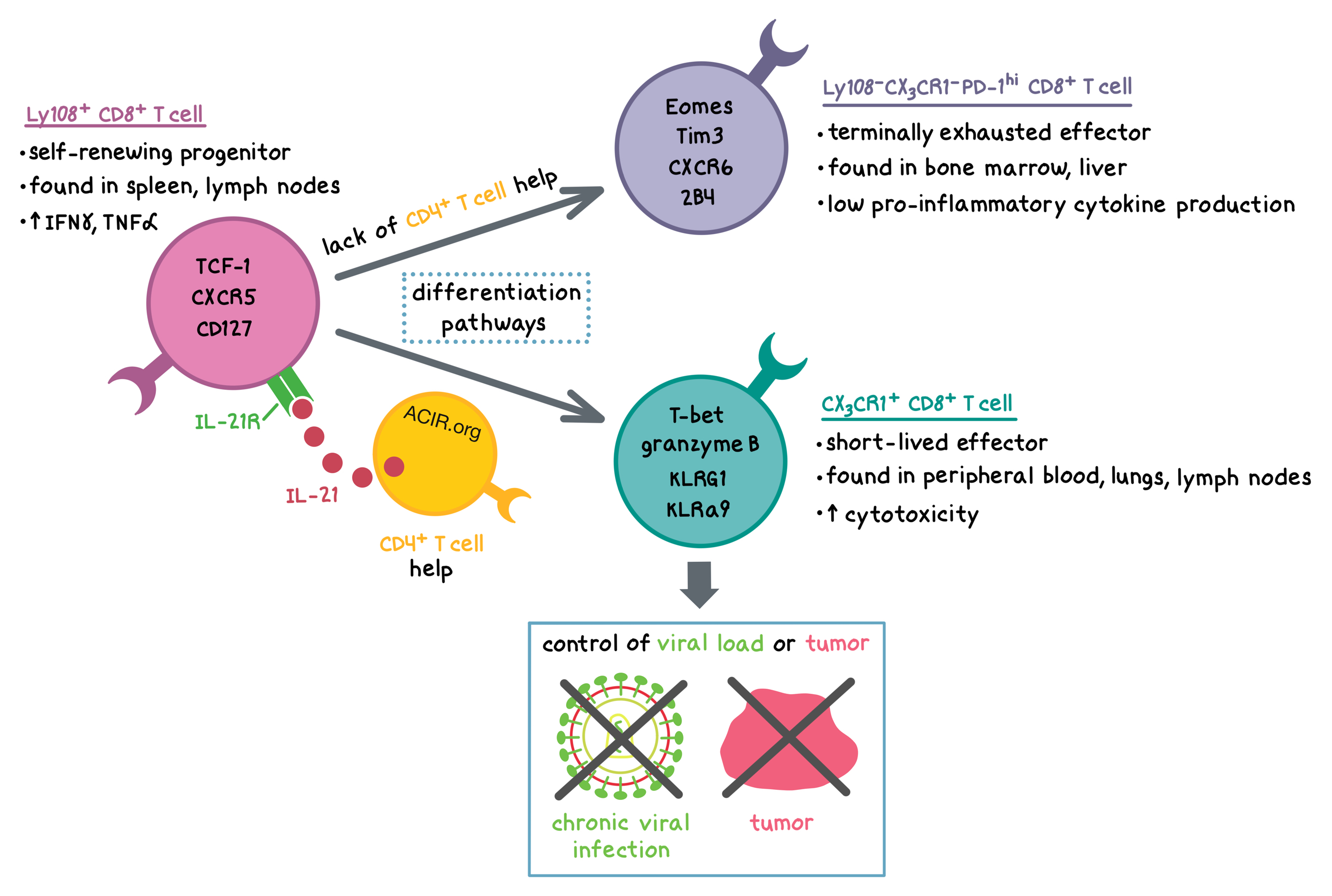
- The Slamf6+ subset (also found at Day 8) appeared to be a self-renewing progenitor population marked by expression of Tcf7, Id3, Icos, and Il7r, as well as an increased expression of Batf3, a transcription factor that is a critical transducer of IL-21 signaling in CD8+ T cells. This subset contained the previously identified CXCR5+TCF-1+ progenitor cells.
- The Pdcd1+ subset appeared to comprise terminally exhausted cells with a high expression of Gzma and Gzmb, but also a high expression of inhibitory receptors such as CD244 (encoding 2B4), Cd160, Lag3, Havcr2 (encoding Tim3), and Pdcd1 (encoding PD-1). This subset was also distinguished by the expression of Cxcr6 and Cd7, as well as transcription factors Nr4a2, Maf, and Eomes.
- The Cx3cr1+ subset appeared to have a profile similar to short-lived effectors in acute infections, characterized by a high expression of killer lectin-like receptors (Klre1, Klra9, Klrd1, and Klrg1) as well as transcription factors Tbx21 (encoding T-bet), Zeb2, Klf2, Id2, and Runx1.
An effector cluster found at Day 8 and the two unique effector-related Day 30 clusters (Pdcd1+ and Cx3cr1+; terminally exhausted and short-lived, respectively) were highly divergent from each other. However, Day 8 effectors did share expression of some genes with each of the two Day 30 clusters, indicating that by day 8, effector CD8+ T cells began to acquire characteristics of the subsets seen at day 30.
To confirm these results at the protein level and to determine tissue distribution, the researchers compared surface marker expression of the chemokine receptor CX3CR1 and Ly108 (encoded by Slamf6) among LCMV-specific CD8+ T cells in mice infected with LCMV Cl13. The Ly108+ subset (representing the Slamf6+ cluster) was mostly found in secondary lymphoid tissues (i.e. spleen, lymph nodes) and displayed a high expression of CXCR5, CD127 (encoded by Il7r), and TCF-1, as well an enhanced capacity for production of IFNγ and TNFα upon peptide stimulation. The Ly108-CX3CR1- subset (representing the Pdcd1+ cluster) was enriched in the bone marrow and liver and had an increased expression of CXCR6, Eomes, PD-1, Tim3, and 2B4. This subset was characterized by the lowest potential for pro-inflammatory cytokine production among the three analyzed subsets, consistent with exhaustion. The CX3CR1+ subset (representing the Cx3cr1+ cluster) was found predominantly in peripheral blood, lungs, and inguinal lymph nodes, and had an increased expression of KLRG1, KLRa9, T-bet, and granzyme B, as well as enhanced cytotoxicity ex vivo against peptide-pulsed target cells. Thus, the surface marker expression data were consistent with the scRNAseq gene expression results.
To determine cell differentiation trajectories, Zander and Schauder et al. applied a machine learning algorithm (Monocle 2) to the scRNAseq data. The algorithm predicted that Slamf6+ cells could terminally differentiate into either Pdcd1+ or Cx3cr1+ cells. Consistent with the algorithm predictions, adoptively transferred Ly108+CD8+ T cells gave rise to the Ly108-CX3CR1-PD-1hi and CX3CR1+ subsets in vivo. Furthermore, when all three subsets were adoptively transferred, Ly108+CD8+ T cells expanded much more than the other two subsets in the blood, spleen, and liver, and had an increased expression of Ki67. Together, these results further support the notion that the Ly108+CD8+ T cells have the self-renewing progenitor phenotype. Interestingly, while the majority of transferred CX3CR1+ cells maintained their phenotype, indicating terminal differentiation status, 40-50% of transferred Ly108-CX3CR1-PD-1hi cells acquired CX3CR1 and T-bet expression, suggesting some heterogeneity with respect to fate determination.
Given previous knowledge that CD4+ T cells play a role in sustaining CD8+ T cell responses during chronic viral infection, that sustained responses rely on CD8+ T cell-intrinsic IL-21R signaling, and that CD4+ T cells have previously been identified as a major source of IL-21, Zander and Schauder et al. explored mechanistically how CD4+ T cells play a role in CD8+ T cell differentiation during persistent viral infection. Consistent with previous observations, over 90% of IL-21-producing lymphocytes in the spleen during LCMV Cl13 infection were CD4+ T cells. Using several types of experiments, including transient depletion, adoptive cell transfer, and bone marrow chimeras, the researchers demonstrated that mechanistically, CD4+ T cell-derived IL-21 played a critical role in facilitating the formation of cytotoxic CX3CR1+CD8+ T cells, which were required for controlling viral replication during persistent viral infection in vivo. CD4+ T cell-derived IL-21 most likely promoted the differentiation of Ly108+CD8+ T cells toward CX3CR1+ cells and suppressed differentiation toward the exhausted Ly108-CX3CR1-PD-1hi phenotype. Furthermore, in an attempt to at least partially reverse T cell exhaustion, the researchers treated mice with PD-L1 blockade and found that although PD-L1 blockade reduced the viral load in mice with intact CD4+ T cells, it failed to do so in mice depleted of CD4+ T cells. Mechanistically, in the absence of CD4+ T cell help, PD-L1 blockade could not rescue the formation of the CX3CR1+CD8+ T cell subset.
Finally, the researchers examined whether tumor-infiltrating CD8+ T cells (TILs) follow a differentiation path similar to the virus-specific cells in a chronic viral infection. In B16F10 melanoma tumors, the majority of TILs were Ly108-CX3CR1-PD-1hi, consistent with a high level of exhaustion. Very few Ly108+ TILs were detected in the tumor. Adoptive transfer of tumor-specific IL-21+CD4+ T cells (with a Th17 phenotype) into B16F10 tumor-bearing mice resulted in a ~2-fold increase (compared to transfer of comparable IL-21-CD4+ T cells) in tumor-infiltrating CX3CR1+CD8+ T cells and enhanced tumor control. IL-21+CD4+ helper T cell response positively correlated with the proportion of CX3CR1+CD8+ TILs and inversely correlated with tumor burden. These effects were dependent on CD8+ T cell-intrinsic IL-21R signaling.
Overall, Zander and Schauder et al. demonstrated that in the context of a chronic viral infection or cancer, CD4+ T cell help – in the form of IL-21 production – was required for the formation of the cytotoxic CX3CR1+CD8+ T cell subset, which was responsible for the control of viral load or tumor, respectively. Furthermore, the presence of CD4+ T cell help was required for the efficacy of PD-L1 blockade. The results of this study elucidate a mechanistic pathway that could play a role in improving the efficacy of cancer-targeting immunotherapies.
by Anna Scherer
Meet the Researcher
First co-author Ryan Zander answered our questions this week.

What prompted you to tackle this research question?
Recent work in the field has indicated that CD8+ T cells responding to chronic viral infection can be broadly compartmentalized into two major subsets, with a CXCR5hiTCF-1hi subset serving as a self-renewing progenitor population that can give rise to a more terminally exhausted CXCR5loTCF-1lo subset. Similar progenitor-like and exhausted CD8+ T cell subsets have also been observed during many different types of cancer. These observations led us to ask whether additional heterogeneity exists among CD8+ T cells responding to persistent infection or tumorigenesis, and whether CD4+ T cell help regulates this multifaceted process of CD8+ T cell differentiation.
What was the most surprising finding of this study for you?
I think that the most surprising finding of this study was the identification of a transcriptionally distinct CD8+ T cell subset that displays potent cytolytic activity, and whose development is wholly dependent on CD4+ T cell help. Although CD4+ T cell help has long been known to be important for sustaining CD8+ T cell responses, the underlying mechanisms by which this occurs have remained unclear. Our discovery that a lack of CD4+ T cell help largely precludes the differentiation of a functional cytotoxic CD8+ T cell subset is really quite striking and has important implications for immunotherapy.
What was the coolest thing you’ve learned (about) recently outside of work?
A recent Snapple fact that I found to be quite entertaining is that flamingos are actually born with grey feathers that then gradually turn pink due to a dye that they obtain from their diet of shrimp.



