In November, the ACIR team attended the SITC Annual Meeting 2021. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
T cell therapies
Crystal Mackall
David O’Malley
Alena Gros
Cassian Yee
Tumor microenvironment and tumor immunology
Lisa M. Coussens
Christine Moussion
Juan R. Cubillos-Ruiz
Giacomo Oliveira
Ellen Puré
Jorge Mansilla-Soto
Checkpoint blockade and in situ therapies
Antoni Ribas
James W. Welsh
Daniel J. McGrail
Craig Slingluff
Paolo A. Ascierto
Immunotherapy approaches
Marcela V. Maus
Sriram Sathyanarayanan
Dimitris Skokos
Robert D. Schreiber
T cell therapies
CAR-T Cells for Solid Tumors: Charting a Path Forward- Crystal Mackall, MD – Stanford University
As the recipient of the 2021 Richard V. Smalley Memorial Award, Crystal Mackall discussed the potential to use CAR T cells for the treatment of solid cancers, and the hurdles that must be overcome in order to do so effectively. Noting previous research, which showed that high levels of antigen density were required for CAR T cells to be effective, Mackall described how what could be seen as a hurdle in some settings, could actually serve as a therapeutic window in others. If CAR T cells only respond to antigens that are present at high densities, then target antigens may not need to be entirely exclusive to the tumor. Recent findings from an early biopsy program for diffuse intrinsic pontine glioma (DIPG), a lethal childhood cancer with no treatment options beyond radiotherapy, revealed high expression of GD2, a known tumor antigen with limited expression on normal tissues. This finding prompted Mackall and colleagues to revive work on a previously developed GD2-targeted CAR T cell, which had struggled with tonic signaling problems. Switching out the CD28 domain for a 4-1BB domain improved the functionality of these cells, which showed efficacy in an orthotopic xenograft mouse model of H3K27M DIPG brain tumors, though some mice were lost early due to toxicity that was unrelated to on-target neurotoxicity. In further efforts to optimize the GD2 CAR T cells, Mackall and her team manufactured them in the presence of Dasatinib, a potent inhibitor of CARs that diminishes exhaustion and improves functionality by turning off tonic signaling; Dasatinib can be washed out prior to transfer. An iCas9 suicide switch that would allow CAR T cells to be shut down if necessary was also incorporated. Moving into Phase 1 clinical trials, additional precautions were taken to manage possible adverse events, including the implantation of an Ommaya catheter to monitor and relieve intracranial pressure. Patients with DIPG and spinal diffuse midline glioma (DMG) with a proven K27M genetic mutation that would cause them to express GD2 were treated with GD2-targeted CAR T cells, and while this study is still ongoing, several patients have shown impressive tumor volume reduction. Importantly, several patients have also experienced significant improvement in tumor-related neurological symptoms, which has improved their quality of life. Some patients have experienced worsening of existing symptoms early on, likely due to swelling of the tumor (coined as TIAN: Tumor Inflammation-Associated Neurotoxicity), but this effect was transient and manageable. While this study was underway, preclinical work on another brain tumor antigen-targeted CAR T cell therapy (B7H3 CAR) showed that intracerebroventricular (ICV) infusion of CAR T cells was more potent than systemic infusion. Incorporating this research into their trial, Mackall and colleagues began delivering GD2 CAR T cells ICV in patients who eventually progressed following their initial IV dose of GD2 CAR T cells. In addition to inducing responses, ICV delivery did not require lymphodepletion, and T cells were found circulating in peripheral blood. Thus far, 10 out of 11 treated patients have experienced clinical or radiographic benefits, with up to 99% reduction in tumor volume. Early evidence suggests effective expansion and persistence of GD2 CAR T cells and infiltration into tumors. Interestingly, ICV delivery also induced infiltration of immune-activating myeloid cells early after infusion, while other delivery methods favored immune-suppressive myeloid subsets. This study is a major step forward for CAR T cells in solid tumor settings, but according to Mackall, it is just the tip of the iceberg.
Phase 2 efficacy and safety of autologous tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab in immune checkpoint inhibitor-naïve patients with advanced cancers- David O’Malley – OSU Medical Center
One-time adoptive cell therapy utilizing the tumor-infiltrating lymphocyte (TIL) products lifileucel (LN-144) or LN-145 (hereafter LN-144/145) in patients with advanced cancer who failed treatment with immune checkpoint inhibitors (ICI) has demonstrated encouraging efficacy with acceptable safety. To provide an early-line treatment that can improve rates and depths of responses, David O’Malley and colleagues explored a combination of TIL therapy and pembrolizumab in patients with ICI-naive melanoma, HNSCC, or cervical cancer. O’Malley reported results from two ongoing Phase 2, multicenter studies (IOV-COM-202 and C-145-04) of LN-144/145 and pembrolizumab in patients with anti-PD-1/PD-L1-naive solid tumors (melanoma [n=10], HNSCC [n=18], or ovarian cancer [n=14]). All patients enrolled had a high tumor burden at baseline and all patients in the cervical cancer cohort had distant metastases at the time of study entry. LN-144/145 was centrally manufactured from a tumor lesion at least 1.5 cm in diameter within 22 days, and cryopreserved. Patients had to have at least one additional tumor lesion for RECIST response assessment. Following tumor resection for TIL manufacturing, the patients received one dose of pembrolizumab and then underwent non-myeloablative lymphodepletion (NMA-LD) prior to a one-time infusion with LN-144/145. Treatment was then followed by up to 6 doses of IL-2, given every 8-12 hours after the completion of TIL infusion, and continued with pembrolizumab every 3 or 6 weeks (depending on the dose) for up to two years. Treatment-emergent adverse events were observed prior to or within the first two weeks after TIL infusion in more than 30% of patients, and were consistent with advanced disease and known safety profiles of pembrolizumab, NMA-LD, and IL-2. Five grade 5 events were reported and evaluated as unlikely to be related to TIL or pembrolizumab administration. At a median follow-up of 12 months (melanoma) or 8 months (HNSCC and cervical cancer), nearly all efficacy-evaluable patients (86-100%) experienced a reduction in tumor burden. Many responses deepened over time, and were durable and ongoing at data cutoff. Objective responses were observed in 60% of patients with melanoma (including a 30% complete response rate), 39% of patients with HNSCC, and 57% of patients with cervical cancer. In conclusion, TIL therapy with LN-144/145 demonstrated efficacy and safety in multiple solid tumor types and lines of therapy, both as a monotherapy and in combination with ICI, and warrants continued investigation.
Biomarkers of Exhaustion Guide the Identification of Tumor-Reactive TIL and Peripheral Blood Lymphocytes- Alena Gros, PhD – Vall d’Hebron Institute of Oncology
Alena Gros’s talk focused on the identification and characterization of tumor-reactive lymphocytes, which are relevant in a variety of immunotherapeutic settings, from TIL therapy, to checkpoint blockade, to personalized T cell therapies. Expansion of TIL ex vivo with IL-2 has served as a strong source for tumor-reactive lymphocytes, but a number of factors influence the detection of tumor-reactive lymphocytes, including the tumor type and its expression of MHC, and the functional and differentiation states of T cells. To address the variability of detection of tumor-reactive lymphocytes, Gros and colleagues set out in search of biomarkers to guide the identification process in a way that is reliable and generalizable across tumor types. Previous studies have identified numerous biomarkers of possible tumor reactivity, including PD-1, LAG3, TIM3, CD103, CD39, and some combinations of markers on CD8+ T cells; similar, though less consistent observations have been noted on CD4+ T cells. Using endometrial cancer as their model, Gros and her team looked for tumor-reactive TILs among 47 primary tumor samples across a range of molecular subtypes (POLEmut, MSI, copy number high [CNH], and copy number low [CNL]) with varying degrees of mutation and prognosis. While POLEmut samples showed the greatest CD3 infiltration, it was highly variable, and fairly high across subtypes, and did not correlate with prognosis. Looking at TILs based on PD-1 expression (PD-1hi, PD-1dim, or PD-1-), the researchers found that PD-1hi cells frequently co-expressed CD39, CD103, and TIM3. Using tumor cell lines generated from 4 patients, the researchers observed high numbers of tumor-reactive T cells within PD-1hiCD39+CD8+ TILs. The combination of PD-1 and CD39 had the greatest predictive value for the identification of tumor-reactive lymphocytes, and these biomarkers further correlated with prognosis. Interestingly, these cells expressed high levels of CXCL13, suggesting that even though they were in some ways dysfunctional, they still produced factors that might influence the TME. Similar, though less consistent results were observed when looking at CD4+ T cells, suggesting more diverse T cell subsets. In another project, Gros and colleagues looked into the potential use of these biomarkers to isolate tumor-reactive lymphocytes from peripheral blood. They had previously shown that circulating PD-1hi CD8+ and CD4+ T cells behave differently compared to similar populations among TILs, and are much lower in frequency. Sorting peripheral blood T cells from patients with diverse tumor types based on expression of PD-1 and other biomarkers (e.g., CD27, HLA-DR, CD38, CD39), the researchers identified tumor-reactive lymphocytes, including those reactive to neoantigens and cancer germline antigens, in 8 out of 10 patients. Again, the combined expression of PD-1 and CD39 had the highest enrichment for tumor-reactive lymphocytes, despite the fact that these cells were rare. Importantly, this method of sorting offered greater detection of neoantigen-reactive lymphocytes compared to TIL expansion in IL-2, offering a strategy for identifying patient-specific tumor-reactive TIL that is less invasive and more accessible than tumor biopsy or tumor resection.
Endogenous T cells- Cassian Yee, MD – The University of Texas MD Anderson Cancer Center
Cassian Yee’s talk was focused on three key aspects that define the efficacy of cellular therapies using peripheral antigen-specific T cell clones; persistence, antigen spread, and tumor microenvironment (TME) modulation. First in a trial in which patients were treated with NY-ESO-1-reactive CD4+ T cell clones without prior lymphodepletion, and then in a trial with gp100 in melanoma patients, patients with a complete response had long-term persistence of CD8+ T cells, while patients with progressive disease had very short-term persistence. Persisting cells had a central memory (Tcm) phenotype. Yee and colleagues found that when cells were treated with IL-21 during the priming phase, antigen-stimulated naive T cells would upregulate CD28 and CD127, suggestive of the Tcm phenotype. In a trial in patients with AML, WT1-specific T cells primed with IL-21 during culture led to Tcm cells that would persist and were associated with a durable clinical response, while T cells that did not undergo this priming were associated with a short-term response. When looking into the IL-21 signaling in those T cells, the researchers found that pSTAT3 binds to the promoter of CD28, resulting in CD28 upregulation. The addition of IL-12 to effector T cells, TILs, or CAR T cells did not affect CD28 expression, but reprogramming of these cells to upregulate CD28 was possible using an HDAC inhibitor. Another important aspect of therapy response is antigen spreading. Tumors of complete responders had a heterogeneous expression of the antigen target, yet were completely cleared, suggestive of antigen spread. This was confirmed in a study using MART-1-targeting CD8+ T cells, in which treatment with these cells induced antigen-specific T cells to multiple other targets in responders. Finally, Yee discussed the potential for combination strategies with this cellular therapy. Since this strategy does not require any conditioning chemotherapy or high-dose IL-2, there is a low risk of toxicities, and it can therefore be readily combined with other therapies that modulate the TME to achieve more complete responses.
Back to Top
Tumor microenvironment and tumor immunology
Neutralizing protumor inflammation: Lessons Learned from Preclinical Mouse Models- Lisa M. Coussens, PhD - Oregon Health & Science University
In her Keynote address, Lisa Coussens summarized and updated her decade-long efforts to understand the dynamic immune contexture in cancer, and identify actionable vulnerabilities and biomarkers. Beginning with human tissue samples, immunohistochemistry and multi-color flow cytometry were used to analyze the immune composition of normal and diseased tissue. This revealed that although different from normal tissues, tumor-containing tissues were more similar to normal tissues than to tumors across tissue types, demonstrating tissue specificity. To begin to understand the origins of these immune contextures, Coussens turned to genetically engineered mouse models (GEMM), in which both pre-malignant and malignant tumors of three different histologies could be examined, and uncovered an important role for Th2-polarizing macrophages and monocytes, the T or B cell drivers, and the mediators of those immune cells. Genetic knockouts of key cells/mediators slowed tumor appearance and converted the tumor microenvironment (TME) to a more antitumor Th1 milieu. Direct administration of myeloid-targeted antibodies or other inhibitors in pre-malignant disease slowed tumor appearance with similar TME changes, while treatment of advanced tumors induced TME changes, but had minimal impact on tumor progression. Combination therapy with chemotherapy or radiation rescued tumor control of these treatments in late tumors, and helped identify the underlying myeloid “vulnerabilities” and important positive mediators of effective T cells. Tumors with low myeloid infiltration had a much higher fraction of early effector PD1+EOMES- T cells, and lower presence of PD1+EOMES+ dysfunctional T cells compared to tumors with a high myeloid TME. Analysis of human tumors showed a negative correlation between myeloid cell infiltration and relapse-free or overall survival, providing important confirmation of the mouse results. Despite these encouraging results, initial studies of various myeloid cell modulators in combination with other therapies did not lead to clinical improvements, despite favorable TME changes, suggesting that improvement of T cell number/phenotype may be necessary. Analysis of human mesothelioma recapitulated the Th2 myeloid-induced TME (with PD-L1+CD206+ macrophages) and showed the repressive effect of these myeloid cells on T cells. Coussens hypothesized that PD-L1+ macrophages may inhibit the efficacy of anti-PD-L1/PD-1 therapy in mesothelioma. A mouse model of mesothelioma allowed evaluation of both primary and lung metastatic tumors and manipulation. CSFR-1 inhibition combined with chemotherapy reduced mesothelioma primary tumor growth, but was not further improved with anti-PD-L1 therapy. However, while the CSFR-1 and chemotherapy combination had no direct effect on metastases, control of metastatic lesions was significantly improved by the addition of anti-PD-L1. All of these effects were CD4+ and CD8+ T cell-dependent, and T cells showed signs of activation, proliferation, cytotoxicity, and TCR clonal expansion. Given the known importance of TCF1+ memory stem cells, Coussens asked if TCF1+ cells expanded following these therapies, which indeed was the case. Finally, to understand the mechanism behind this expansion, gene expression analysis of responding tumors in the mouse identified, among others, increased expression of IL-27, a cytokine recently implicated in self-renewal of CD8+ T cells during viral infection. Expression of IL-27 and IL27Rα was confirmed in human mesothelioma samples, and neutralization of IL-27 in the mesothelioma mouse model blocked antitumor responses. TCGA data showed a correlation between IL-27 expression and T cell levels in both mesothelioma and melanoma. Further, both IL-27 levels and macrophage levels correlated positively and negatively, respectively, with survival in a study with anti-PD-1 therapy. All these results were confirmed in the YUMM/YUMMER melanoma model, with anti-CSF therapy rescuing ineffective anti-PD-L1 therapy, whereby rescue was dependent on IL-27. Overall, Coussens showed the power of deep analysis of immune biology within the TME in both human and mouse systems in parallel. She also revealed the importance of tipping the balance of pro- versus anti-tumorigenic macrophages, and highlighted the importance of a multi-nodal approach to overcome the complexity of tumor immunobiology.
Deciphering the spatiotemporal control of tumor immune phenotype- Christine Moussion, PhD - Genentech
Tumors can be categorized by histochemistry into three different immune phenotypes: inflamed, immune excluded, and immune desert. A response to immunotherapy is enriched in tumors with an inflamed phenotype, however little is known about the development, heterogeneity, and dynamics of the three immune phenotypes. To address this question, Christine Moussion and her team developed a high-throughput live-imaging technology, called Skin Tumor Array by Micro-Poration (STAMP), to explore the spatial organization and functions of immune cells controlling tumor immune phenotypes. A laser was used to create an array of small pores in the skin of the ear of RAG2-/- mice reconstituted with TdTomato+ wild-type T cells. GFP-expressing clonal tumor cells were then seeded inside these laser-created micropores. Using live-imaging and machine learning-based image analysis, Moussion and colleagues could follow tumor and T cell dynamics and locations, and found that all three immune phenotypes, including a fourth termed “rejected”, existed in the same array, and that about 30% of the tumors were spontaneously rejected by antigen-specific CD8+ T cells. Heterogeneity in immune phenotype was also observed in a lung metastases model, indicating that the niche at tumor seeding is important in determining the immune phenotype. Early tumor biopsies and scRNAseq and TCRseq of each immune phenotype revealed that even though the immune desert phenotype had low T cell numbers, desert, inflamed, excluded, and rejected phenotypes had similar abundance of T cell subsets and a shared T cell repertoire, suggesting that the T cell repertoire does not dictate tumor immune phenotype and tumor fate. Increased transcription, translation, and mitochondrial biogenesis in inflamed tumors made Moussion and colleagues wonder whether T cell function would be a better marker. The researchers used KPP tumors expressing the calcium sensor GCaMP6 to read T cell attack of tumor cells as a green flash caused by Ca2+ influx. Although inflamed and excluded tumors show similar total T cell abundance, inflamed tumors had a higher flashing index and a slower tumor growth rate than excluded tumors, indicating that an immune inflamed environment was probably a nurturing niche for CD8+ T cell function. To understand what controls generation of the inflamed environment, Moussion and her team used single cell sequencing of multiple tumors and found that the composition of myeloid cells across tumor immune phenotypes was different. Monocytes and mo-DCs were increased in inflamed and rejected tumors, and TREM2+ macrophages and a particular subset of DC1 (DC1:N) were enriched in immune desert tumors. Cell depletion experiments early after tumor seeding indicated that Ly6C+ or Gr1+ cells played a critical role in recruiting and/or retaining T cells at the tumor site, allowing the tumor to shift to an immune desert state. Treating mice with anti-PD-L1 or a combination of anti-PD-L1 and anti-TGFβ increased the number of T cells recruited to KPP STAMP tumors, but only the combination therapy strongly promoted the transition to an inflamed immune phenotype and induced significant tumor regression. Interestingly, Moussion and her team observed that even tumors in the untreated control group could transition between different immune phenotypes. Moussion concluded that the main drivers of the tumor immune phenotype include the tumor (cell of origin, tumor genetics), the host (germ line genetic, systemic immunity), and the state of the tumor niche (time and space component controlled by myeloid/stroma).
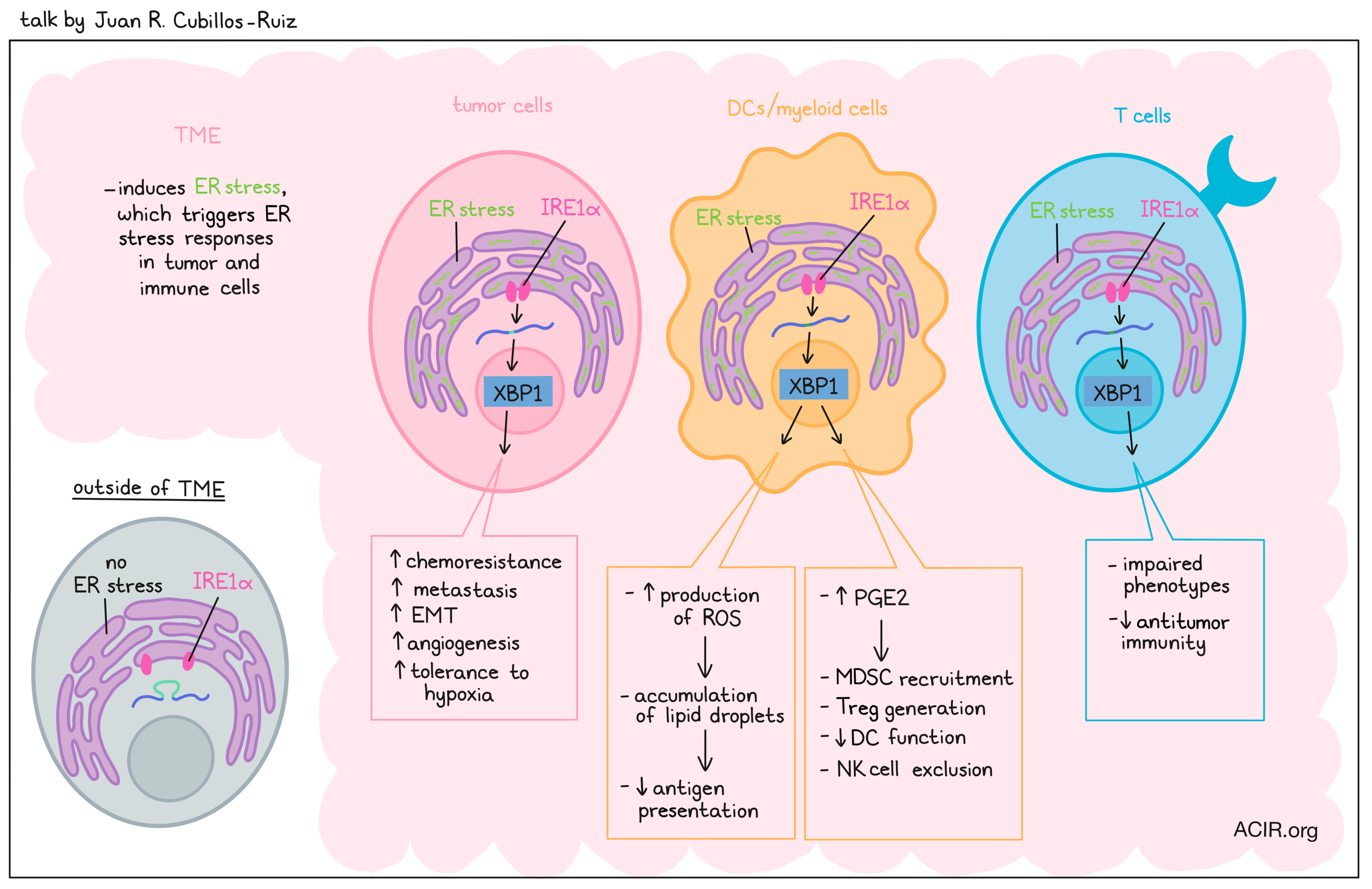
Immunoregulatory Programs Controlled by ER Stress in Cancer- Juan R. Cubillos-Ruiz, PhD – Weill Cornell Medicine
Juan Cubillos-Ruiz discussed the potential of targeting immunosuppressive features of the TME as a category of cancer immunotherapy, focusing on factors that affect infiltrating immune cells. One such factor is ER stress, which can affect protein folding, leading to an accumulation of unfolded or misfolded proteins. To cope with this, ER stress responses may be induced in cells, which can lead to autophagy, cellular reprogramming and adaptation, or cell death. The mechanism for the ER stress response involves 3 major sensors: ATF6, IRE1α, and PERK. The magnitude and duration of environmental stressors can affect the progression of the ER stress response, with mild but persistent stress promoting pro-tumoral adaptations. Under ER stress, IRE1α dimerizes, processes inactive XBP1 RNA to active XBP1s RNA, and induces production of the transcription factor XBP1. XBP1 initiates programs to mitigate the accumulation of unfolded or misfolded proteins, inducing chaperones, foldases, ERAD (quality control), autophagy, and amino acid metabolism. Cancer cells thrive under these conditions, exploiting pathways that allow for chemoresistance, metastasis, EMT, angiogenesis, and tolerance to hypoxia. Investigating how immune cells respond to ER stress, Cubillos-Ruiz and colleagues examined ovarian tumors, where DCs do not effectively present antigens, and instead promote immunosuppression in the TME. DCs from this setting showed hyperactive IRE1α/XBP1 pathways, and selective deletion studies showed that DCs were, in fact, exploiting ER stress to promote tumor progression and metastasis. Further investigation of this mechanism showed that DCs under ER stress produced more reactive oxygen species, driving the accumulation of lipid droplets that prevent effective antigen presentation. Further, XBP1 was found to bind to promoter regions of two key enzymes that promote prostaglandin biosynthesis under ER stress, as they are required for the conversion of arachidonic acid to PGE2. The excess production of PGE2 induced immunosuppression in the TME by recruiting MDSCs, promoting Treg generation, preventing DC function, and promoting NK cell exclusion. When XBP1 was silenced in vivo using nanoparticles, DCs showed enhanced antigen presentation, and T cells showed increased function and memory cell generation, leading to enhanced antitumor responses and longer survival. Similar results were observed in a variety of myeloid cells under ER stress. Finally, Cubillos-Ruiz investigated the effect of ER stress on T cells, and found that IRE1α/XBP1 pathways were again upregulated. Deleting this pathway in normal T cells in naive mice had no effect on normal T cell development or differentiation, but in the tumor microenvironment, infiltrating TILs showed improved T cell phenotypes that enhanced antitumor immunity and survival in tumor-bearing mice. Given that the IRE1α/XBP1 pathway appears to hinder antitumor immunity in a number of ways across a wide variety of cell types, Cubillos-Ruiz and his team are eagerly investigating strategies to inhibit this pathway, enhance immunotherapy, and improve outcomes for patients with cancer.
Landscape of helper and regulatory CD4+ T cells in melanoma- Giacomo Oliveira, PhD – Dana-Farber Cancer Institute
CD4+ T cells play a central role in pro-tumor and anti-tumor immunity, which correlates with their phenotypic state, but how such cells are elicited, and the relationship between T cell phenotype, antigen-specificity, and the mechanism of HLA class II epitope presentation (indirect via APCs; direct via inflamed tumor cell induction of HLA class II; aberrant HLA class II expression characteristic of a few tumor types, such as seen in 10-30% of melanomas) need to be determined. To address these questions, Giacomo Oliveira and his team used single-cell transcriptome and TCR sequencing with TCR reconstruction to characterize the CD4+ T cell repertoire in 4 melanoma tumor samples. Tumor cell lines were prepared from each melanoma tumor sample, and flow cytometry showed that two of the tumors did not express HLA class II, while two demonstrated stable, constitutive HLA class II expression. Nearly 28,000 CD4+ T cells from the four tumors were sequenced, which could be mapped to three major categories: memory, regulatory, and exhausted (both terminally exhausted as well as progenitor exhausted cells with features of follicular helper cells). TCR clonality (indicative of clone-specific expansion) was highest in the regulatory and exhausted compartments. When analyzed by patient, the most dominant clonotypes were in the memory and exhausted compartments in patients who did not express HLA class II, while the regulatory compartment dominated those patients expressing HLA class II. More than 130 TCRs (linked through sequencing to T cell phenotype) were reconstructed from three patients across these compartments to determine antigen specificity by multiplex measurement of CD137 induction following stimulation with various antigens. While TCRs from memory cells for all three patients were mostly devoid of tumor cell recognition, those from terminally exhausted cells from all three patients strongly recognized cognate tumors. Interestingly, these TCRs on CD4+ T cells from the patient who did not express HLA class II recognized tumor-associated antigens presented on HLA class I. Following direct presentation by tumor cells, TCRs from regulatory cells only recognized the two tumors expressing HLA class II, suggesting that HLA class II-expressing tumors can directly activate and expand Tregs in these tumors. Deorphanization of these regulatory TCRs showed that these predominantly recognized neoantigens. In addition, Oliveria studied indirect antitumor recognition via APCs and found that the majority of T cells expanded via this mechanism also had a Treg phenotype. Finally, to determine if not only HLA class II expression by the tumor, but also the number of neoantigens expressed by the cell contribute to this predominance of regulatory T cells in these tumors, the relationship of tumor mutation burden (TMB) and HLA class II expression was evaluated in a larger data set. Extremely high or high TMB were more common in tumors expressing HLA class II, supporting this possibility.
Flanking Solid Tumors via Immune-Mediated Disruption of Desmoplastic Stroma- Ellen Puré, PhD – University of Pennsylvania
One of the reasons that immunotherapies are not effective in many patients with solid tumors is due to immunosuppressive effects of the tumor stroma. Ellen Puré discussed work on CAR T cell therapy targeting the desmoplastic stroma in pancreatic cancer. This cancer type has a poor prognosis, responds poorly to checkpoint blockade, and is characterized by poor infiltration of TILs, a fibroinfammatory response, and matrix remodeling that results in desmoplasia. In addition, cancer-associated fibroblasts (CAFs) are often associated with treatment failure and immunosuppression. However, Puré and others’ research has shown that some CAFs are tumor-restraining while others are tumor-promoting. This heterogeneity in the CAF pool is a major obstacle to therapeutically targeting CAFs. The different CAF populations can be distinguished by their expression of fibroblast activation protein (FAP) and smooth muscle actin (SMA). Given that FAP expression correlates with worse clinical outcomes in patients with pancreatic cancer, the researchers developed a CAR T cell targeting FAP to selectively reduce or eliminate the FAP-expressing CAFs. These CAR T cells infiltrated solid pancreatic mouse tumors, with early infiltration in the stroma that resulted in the killing of FAP+ stromal cells, dissolution of the collagen-rich desmoplastic matrix, and eventually, infiltration of CAR T cells into the tumor nest. The therapy inhibited tumor progression, and dosing twice resulted in long-term efficacy. There was an increase in endogenous T and NK cells in those tumors, while there was a decrease in macrophages, DCs, MDSCs, and tumor vasculature. These proof-of-concept data showed that targeting FAP+ cells may be another approach to attacking solid tumors, especially in combination with other immunotherapy treatments. Puré aims to proceed to the clinic with FAP-CAR T cells, and is working on PET imaging using labeled FAP inhibitors to stratify patients and to be able to monitor therapy efficacy.
HLA-independent T cell receptors effectively target low abundance antigens- Jorge Mansilla-Soto, PhD – Memorial Sloan Kettering Cancer Center
Despite the remarkable success of CAR T cell therapy, a significant number of patients experience a disease relapse. Some of these relapses are due to a decrease in target antigen levels, suggesting that CAR T cells require a certain antigen threshold for activation. Since the TCR-CD3 complex is able to efficiently activate T cells even in the presence of a small number of specific MHC-peptide molecules on target cells, Jorge Mansilla-Soto hypothesized that a TCR-based CAR would display an increased sensitivity to low antigen levels. Based on structural models showing that the variable domains of immunoglobulins and TCRs, when bound to target, share a highly similar structure. Mansilla-Soto fused the variable heavy and light regions of an CD19-specific scFv antibody with the constant alpha and beta regions (respectively) of a TCR, to create a HLA-independent TCR (HIT), and targeted the construct to the TRAC locus for uniform expression under the control of the TCRα promoter. The HIT receptor was expressed and functional in T cells. To study the sensitivity of the HIT receptor, Mansilla-Soto and colleagues created NALM6 target cells with varying antigen levels on their cell surface, ranging from 27,000 to approximately 15 molecules per cell. HIT T cells were compared with T cells expressing a 19-28z CAR construct that was previously shown to provide superior antigen sensitivity compared to a 19-BBz CAR construct. The HIT receptor provided increased sensitivity to low antigen levels in vitro and in vivo, and HIT T cells showed increased cytokine production (IFNγ, IL-2, TNFα), signaling, and degranulation, and decreased time to target cell killing, even in response to target cells expressing only very low antigen levels.
Back to Top
Checkpoint blockade and in situ therapies
Igniting the Fire: Intratumoral Therapy to Turn on Immune Response to Cancer- Antoni Ribas, MD, PhD – University of California Los Angeles
According to Antoni Ribas, immunotherapy works because the cancer cell, at some point, becomes an enabler of the immune response. Upon tumor infiltration and antigen encounter, tumor-specific T cells produce IFNγ. Beyond sending cancer cells into growth arrest and death, conserved IFNγ signaling turns on over 500 genes that make the cancer cells more immunogenic by increasing antigen presentation, and change the tumor microenvironment by secretion of chemokines and attraction of more immune cells to the tumor, together leading to an amplification of the antitumor immune response. Recent studies have shown that IFNγ also has long-distance effects on bystander cancer cells that reach beyond the immediate cancer cells recognized by antitumor T cells. Moreover, IFNγ signaling in cancer cells induces the expression of the PD-L1 as a means for the cancer cells to protect themselves from T cell attack. Therapy with anti-PD-1 or anti-PD-L1 checkpoint blockade can release the negative interaction between PD-1 and PD-L1 and let antitumor T cells do their work. However, in many patients, the preexisting immune response is not strong enough, or patients acquire resistance to anti-PD-1/PD-L1. In some cases, Ribas and his team found JAK1 or JAK2 loss of function mutations downstream of the IFNγ-receptor signaling pathway, leading to immune escape. In this case, T cells still recognize the tumor but the IFNγ amplification of the response no longer works, which is why the cancer progresses. Ribas hypothesized that therapy with intratumoral TLR agonists could ignite the “interferon response fire”. Within the tumor microenvironment are other immune cells, including plasmacytoid dendritic cells, which have high levels of certain TLRs, such as TLR9, and produce type I interferons in response to TLR agonist stimulation. Indeed, intratumoral therapy with a TLR9-agonist, together with systemic anti-PD-1 was able to reverse resistance to anti-PD-1 in a preclinical JAK1/2 knockout resistance model, and produced objective responses in 25% of patients with advanced metastatic melanoma who had progressed or had stable disease on anti-PD-1 therapy. Tumor regression was observed in both injected and non-injected tumor lesions. Ribas pointed to a poster presented at SITC by Kirkwood et al. showing that intratumoral injection of a TLR9 agonist also demonstrated single-agent activity in 20% of patients with PD-1 blockade-refractory melanoma, and work from Kalbasi et al. demonstrating that a dsRNA agonist (BO-112) can bypass defective tumor-intrinsic IFNγ signaling and induce MHC class I expression in an IFN-independent manner, re-sensitizing tumor cells to CD8+ T cell killing. Preliminary results of using BO-112 plus pembrolizumab to treat patients with cutaneous or mucosal advanced melanoma progressing on prior pembrolizumab in a Phase 2 trial was presented in a poster at SITC by Marquez-Rodas et al.. In this trial, an objective response rate of 27% was observed. Together, these data support the concept of igniting the fire through intratumoral administration of agents that induce type I interferon production in patients who have a low basal immune response or who are resistant to anti-PD-1/PD-L1 therapy.
Using Low Dose Radiation and Nanoparticles to address PD1 Resistance- James W. Welsh, MD – The University of Texas MD Anderson Cancer Center
Patients with advanced metastatic cancer suffer from a high systemic disease burden with many complex interconnected tumors. James Welsh presented work focused on the use of radiation therapy (RT) to create an in situ vaccine and start the cancer immunity cycle, which can produce a systemic response and affect untreated tumor sites. Welsh made the distinction that not all RT is equal. In a clinical trial in which stage 4 non-small cell lung cancer (NSCLC) was treated with pembrolizumab with or without RT, it was found that those treated with stereotactic RT had a doubling in the response rate, while widefield RT killed lymphocytes and resulted in a reduction of the effects of pembrolizumab. The researchers found that lymphocyte-sparing radiation of one tumor could improve pembrolizumab efficacy. In a subsequent trial, anti-PD-1-resistant patients received RT of one tumor, and 73% of lesions had some clinical response. However, abscopal effects were rare (11%), which is likely due to stroma that limits lymphocyte infiltration in untreated tumors. Welsh and his team aimed to change that through their RadScopal approach, in which one or two tumors are treated with high-dose RT as the “vaccine” to prime an immune response, while all other tumors are treated with low-dose RT to “pull in” lymphocytes. The low-dose RT resulted in increased infiltration of CD4+ and CD8+ T cells and NK cells, induced macrophage polarization from M2 to M1, decreased TGFβ, reduced cancer-associated fibroblasts (CAFs), increased production of cytokines, and induced metabolic reprogramming. Welsh showed examples of two patients who had progressed on anti-PD-1 and anti-4-1BB or high-dose radiation, but then responded to low-dose RT. RadScopal therapy resulted in a response rate of 55% in patients with anti-PD-1-resistant NSCLC. The treatment worked particularly well for liver metastases and was relatively safe. Welsh also combined the strategy with cellular therapy. An example included a patient with metastatic melanoma who progressed on 15 previous therapies and had received a very high dose of engineered TIL without response. However, the patient achieved a complete response following treatment with low-dose RT, likely due to stroma modulation to allow tumor infiltration by TILs. In more recent cases, Welsh applied RT before TIL therapy. The RadScopal approach was further optimized to the pulse RadScopal approach, in which the first priming dose was used to induce innate priming, and a second prime dose a month later was used to induce an adaptive response – like the second dose of a vaccine. This approach is currently combined with anti-PD-1 and anti-CTLA-4 in a trial for metastatic NSCLC. Finally, Welsh discussed the use of hafnium oxide nanoparticles (NBTXR3) to enhance the on-site radiation effect and further improve the immune priming. In a mouse study, NBTXR3 was combined with RadScopal, anti-PD-1, and anti-CTLA-4, which improved outcomes and abscopal responses, increased TCR diversity in the tumor, and produced memory responses. Trials have now started for this combination treatment.
High TMB Fails to Predict Immune Checkpoint Blockade Response in All Cancer Types- Daniel J. McGrail, PhD – The University of Texas MD Anderson Cancer Center
Tumor mutational burden (TMB) has been associated with more immunogenic solid tumors because of a higher load of mutations, and with that, potentially more neoantigens. This became of particular interest when the FDA approved ICB for high TMB (>10 mutations per megabase) cancer types. However, as described by Daniel McGrail, a high neoantigen load is not always correlated with increased T cell infiltration. For context, McGrail pointed out that when looking at T cell infiltration, diseases associated with statistically significant correlation of high baseline T cell infiltration and neoantigen load (Category I) represented only 25% of newly diagnosed cancers, while those with a non-significant correlation (Category II) represented 50% of new cases; the diseases studied in the trial leading to approval of TMB as a biomarker represented only 10% of new cases. To investigate more deeply, McGrail presented data from a meta-analysis on historic cohorts to establish whether baseline TMB is predictive for immune checkpoint blockade (ICB) response. For the meta-analysis, McGrail and colleagues gathered ICB data from 35 cohorts, including 1,500 patients. Data suggested that Category I cancers have a strong CD8 and neoantigen correlation, while Category II cancers do not have a correlation between neoantigens and CD8. High TMB (TMB-H) predicted ICB response in Category I cancers, while this biomarker was not predictive for response in Category II cancers. However, given the low number of samples per cohort for Category II cancers, the analysis was repeated with all cancers within a category pooled together to increase the power. This revealed that Category II cancers were not inherently less responsive than Category I cancers, indicating that this feature reflects a baseline measure of overall response. However, when segregated by TMB in each category, TMB-H predicted ICB response in Category I (Hazard Ratio of 4.2), but failed to predict response for the larger Category II cohort. The researchers then looked at alternative TMB threshold values or continuous TMB, and none of these improved the response prediction in Category II cancers. Therefore, the data suggested that the cancer context may be important when considering TMB as an ICB biomarker. Currently, there are no set standards to generalize TMB scores between different institutions, and technical factors may affect the score – another variable that must be considered. These technical factors include the inclusion of coding vs. non-coding regions and non-silent vs. silent mutations, the variant allele frequency threshold, approaches to account for germline variants, whether primary or metastatic tumors are assessed, and consideration of specific therapy effects.
Intratumoral immune therapy for recurrent breast cancer with polyICLC, and tremelimumab combined with systemic durvalumab- Craig Slingluff, MD – University of Virginia
Craig Slingluff described the lead up to and the results of an innovative trial of multiple immune modulators (LUD2014-001) – intratumoral injection of anti-CTLA-4 (tremelimumab) followed by intratumoral/intramuscular immune adjuvant (pICLC) and systemic anti-PD-L1 (durvalumab) –- in patients with advanced breast cancer. The rationale for the combination included improved safety (due to lower dosing needed for intratumoral anti-CTLA-4), dendritic cell activation and a type I interferon response by pICLC, benefits of combination with anti-PD-L1, and the possibility of creating an in situ vaccine with systemic benefit. The initial trial results had demonstrated safety of a recommended combination dose (RCD) of each agent, and expansion at that RCD in an 8-disease basket trial confirmed safety and showed a signal of activity in the breast cancer cohort. That breast cancer cohort was further expanded to 19 patients in total, and was the focus of Slingluff’s presentation. The 19 patients had a median of 4 prior lines of therapy, including 3 with anti-PD-1/PD-L1. 17 of these patients received all 3 modalities (2 patients only received anti-PD-L1 and pICLC). One patient had a complete response (CR) affecting multiple sites (skin, lymph node, and lung), and multiple patients demonstrated partial response (PR) or stable disease, for an overall response rate (CR + PR) of 26% in this heavily pre-treated population. Comparable frequencies of responses were observed in both triple-negative breast cancer (TNBC, n = 9) as well as non-TNBC (n = 10). For the intent-to-treat population, the median overall survival was 10.8 months, with 37% of the patients still alive at last follow-up (8-46+ months). Multiple favorable correlative changes were noted following immune analysis of the tumor microenvironment, including increased numbers of CD8+ T cells, CD20+ B cells, mature DCs, CD56+ NK cells and macrophages, and increases in antigen-experienced, granzyme B-producing, proliferating (Ki-67+) CD8+ T cells. Increased Ki-67+CD8+ T cells correlated with patients showing an objective response. No changes were noted in markers of immunosuppression (IDO, Arg1, CD39, CD73).
Is Anti-LAG-3 the Next Effective Immunotherapy Treatment? Certainties and Controversies- Paolo A. Ascierto, MD – Istituto Nazionale Tumori IRCCS Fondazione G. Pascale
To improve the efficacy of immune checkpoint blockade (ICB), trials are now expanding to include blockade of LAG3, next to targeting the PD-1/PD-L1 and CTLA-4 pathways. Paolo Ascierto provided an overview of clinical studies assessing the impact of anti-LAG3 combined with other ICB. LAG3 sends an inhibitory signal to T cells upon binding to its ligand, MHC-II, down-modulating T cell proliferation and activation. Expression of LAG3 in tumors has been associated with worse outcomes, and LAG3 was found to be upregulated in patients with renal cell carcinoma and melanoma after anti-PD-1 treatment. Therefore, LAG3 may play a role in therapy resistance. In a trial for solid tumors, anti-LAG3 (relatlimab) was assessed alone or in combination with anti-PD-1 (nivolumab). The combination resulted in higher efficacy and CD4+ and CD8+ memory T cell responses. In patients with melanoma who progressed on previous ICB, an objective response rate (ORR) of 11.5% was reached, and LAG3 expression predicted a better response. A study of neoadjuvant and adjuvant anti-PD-1 with anti-LAG3 in patients with advanced melanoma presented by Rodabe Amaria at ASCO this year showed a 59% pathological complete response rate. The RELATIVITY-047 trial, presented at ESMO this year by Stephen Hodi, combined anti-LAG3 with anti-PD-1 and compared it to anti-PD-1 alone in previously untreated advanced melanoma. The addition of anti-LAG3 improved the median progression-free survival (PFS) by 5.5 months. When these patients were stratified according to LAG3 expression, no difference in effect was seen, suggesting LAG3 expression might not be a good biomarker. The combination also reduced the risk of progression after the following line of therapy. Various trials have now also studied other anti-LAG3 antibodies or soluble LAG3 protein with PD-1/PD-L1 blockade with encouraging results, particularly in metastatic melanoma, and a manageable safety profile. An ongoing trial combines anti-LAG3 with anti-PD-1 and anti-CTLA-4 or an IDO inhibitor in an attempt to further improve outcomes. The role of biomarkers to predict therapy efficacy remains to be defined.
Back to Top
Immunotherapy approaches
Next generation approaches in T cell engineering- Marcela V. Maus, MD, PhD – Massachusetts General Hospital
CAR T cells are a useful tool in immunotherapy, but Marcela Maus and her colleagues work under the hypothesis that CAR T cells need to be “fit for purpose”, as different tumors not only require novel targets, but also present unique obstacles and require different approaches. To address this, the researchers are studying both mechanisms of action and mechanisms of resistance in CAR T cell therapy. In one study, the team developed CAR T cells targeting CD37, as this antigen is expressed widely on lymphomas and leukemias, including some T cell lymphomas and AML. While CD37 is also expressed on a number of non-target immune cells, CD37 CAR T cells induced limited markers related to killing of these cells in vitro. When tested against target cells, CD37-targeted CAR T cells were as effective as CD19-targeted CAR T cells in vitro. In vivo, they induced durable regressions in a number of treated mice, prompting a Phase 1 clinical trial, which has shown early promise; the full results of this will be presented at ASH. In other research focused more on the mechanisms by which CAR T cells kill tumor cells, Maus and colleagues investigated the role of IFNγ and its association with cytokine release syndrome (CRS) and neurological adverse events. Interestingly, they found that in an in vitro setting of CD19 CAR T cells targeting leukemia or lymphoma cells, blocking IFNγ had little to no effect on CAR T cell-mediated killing of target cells. Similar results were observed in mice, where the addition of an IFNγ-blocking antibody had no impact on antitumor efficacy or survival. As a more efficient means of eliminating IFNγ, the researchers used genetic engineering to knock out IFNγ, along with TRAC, in CAR T cells. As with IFNγ blockade, IFNγ knockout had no effect on the antitumor efficacy of CAR T cells. To investigate whether IFNγ knockout impacted toxicity, the researchers had to develop models that could reliably recapitulate CRS in patients. They showed that following incubation with GM-CSF, macrophages from healthy donors that were stimulated with serum from patients who later went on to develop cytokine release syndrome had a cytokine profile comparable to that seen in patients undergoing CRS. The researchers then went on to show that co-culturing serum from leukemia/lymphoma-bearing NSG mice treated with CD19 CAR T cells with GM-CSF-activated macrophages from healthy human donors also replicated that profile. Using this model, Maus and colleagues saw that while wild-type CD19 CAR T cells induced macrophage activation, the IFNγ-knockout CD19 CAR T cells did not, reducing the cytokine profiles for IL-6, IP-10, and MPC-1. Similar results were observed using BCMA-targeted CARs, showing that the results were not CAR target-specific. Interestingly, in a separate study, a CRISPR screen showed that in addition to antigen loss, downregulation of IFNγ receptors acted as a major mechanism of resistance to CAR T cell therapy in glioblastoma. Similarly, in a study of EGFR-targeted CAR T cells in mice bearing EGFR+ brain tumors, IFNγR1 knockout in tumors conferred resistance to CAR T cell therapy, suggesting that IFNγ does play a role in the CAR T cell-mediated antitumor effect in this solid tumor setting. Similar effects were observed in other solid tumor models using CARs targeting various antigens. This research highlights the importance of addressing tumor-specific factors when developing CAR T cell therapies, as the tumor setting can dramatically impact the mechanisms of antitumor immunity.
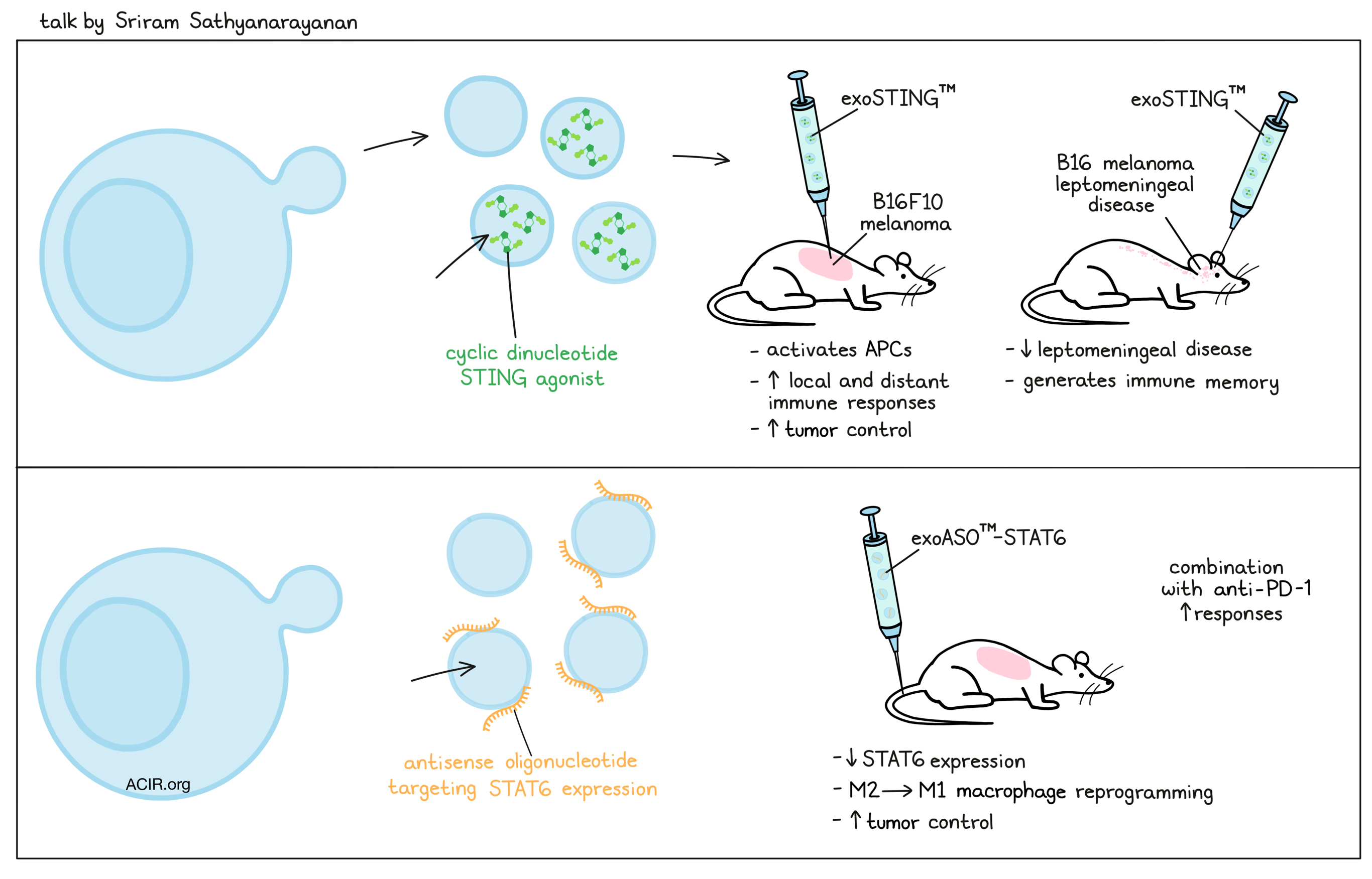
Developing Engineered Exosome Based Therapeutics for Immuno-Oncology- Sriram Sathyanarayanan, PhD – Codiak Biosciences
Turning attention to exosomes, a natural cell-to-cell communication vehicle, Sriram Sathyanarayanan gave an overview of the some of the key biological features of exosomes and then described how exosomes have been engineered and manufactured for two immunotherapeutic applications currently in clinical development. Exosomes are made by all cells as small- to mid-sized nanoparticles, and contain both passenger membrane and luminal cell components (integrins, immune signaling components, intracellular proteins, DNA, and multiple other nucleic acids) as well as specific molecules that are targeted for exosome incorporation (PTGFRN or BASP-1), which can be used to incorporate a variety of payloads. Natural exosomes are safe (trillions are present in each blood transfusion), but heterogeneity and low natural payload incorporation have necessitated engineering and significant clinical manufacturing development to utilize exosomes as a therapy. The first therapeutic application, developed by Sathyanarayanan and his colleagues at Codiak Biosciences, incorporates a proprietary cyclic dinucleotide STING agonist into exosomes (exoSTINGTM). In mouse models of B16F10 melanoma, intratumoral delivery led to uptake by and activation of antigen-presenting cells, stimulating local and distant immune responses. Preclinical results of treating leptomeningeal disease – the spread of tumor cells into the meningeal compartment of the brain – were shown, indicating that intrathecal or intracerebroventricular administration of exoSTINGTM led to significant reduction of tumor growth and improved survival in animals with B16 melanoma brain disease. Moreover, protective immunological memory was generated. ExoSTINGTM is currently being studied in a Phase 1/2 trial. A second application for exosomes discussed by Sathyanarayanan highlighted delivery of a different type of payload – an antisense oligonucleotide that targets STAT6 expression (exoASOTM-STAT6), as STAT6 is a key transcription factor involved in M2 macrophage differentiation and is linked to poor prognosis in hepatocellular carcinoma. Intravenous delivery of exoASOTM-STAT6 resulted in accumulation in the liver and spleen, and in HCC tumors. The exosomes were preferentially taken up by M2, but not M1, macrophages, resulting in a shift in M2 macrophage phenotype to M1 and reprogramming of the immunosuppressive tumor microenvironment. ExoASOTM-STAT6 monotherapy or combination therapy with anti-PD-1 led to improved tumor control in an orthotopic HCC tumor model, supporting clinical testing of this approach.
Human Costimulatory Bispecific Antibodies in Cancer Immunotherapy: Focus in Solid Tumors- Dimitris Skokos, PhD – Regeneron Pharmaceuticals, Inc.
As the immune infiltrate in tumors has a major impact on the success of immunotherapy, therapies can be specifically tailored to address the immune environment. Dimitris Skokos and colleagues at Regeneron developed various building blocks of therapy around different types of bispecific antibodies to create combinatorial flexibility to tailor to different tumor microenvironments (TMEs). In order to improve purification, manufacturing stability, and the final quality of the engineered bispecific antibodies, Skokos and colleagues combined a single “common” light chain to avoid issues with the light chain missparing with two amino acid substitutions in IgG1 (IgG*), to eliminate binding to protein A, and to allow for selective isolation of the bispecific antibody. In addition, the Fc region can be modified to reduce the effector function. CD3 bispecific antibodies bridge a tumor-specific antigen by targeting the TCR/CD3 complex (T cell activation “signal 1”), while CD28 bispecific antibodies mimic the costimulatory “signal 2” by binding to CD28 on T cells. Skokos reported on three such antibodies that are currently tested in Phase 1/2 trials in combination with cemiplimab (anti-PD-1): PSMAxCD28 in prostate cancer, EGFRxCD28 in advanced solid tumors, and MUC16xCD28 in ovarian cancer. The researchers found that PD-1 can exclude CD28 from the immune synapse. PD-1 inhibition circumvents this effect, resulting in enhanced accumulation of CD28 in the synapse. PSMAxCD28 combined with anti-PD-1 increased cytotoxic potency in vitro, and promoted tumor killing without systemic toxicity in vivo. PD-1 alone expanded effector CD8+ T cells in the tumors, while combination therapy expanded CD8+ TILs with a memory-like and less exhausted phenotype (lower expression of PD-1, LAG3, and TIM3). No systemic toxicity was detected in primates for the bispecific, while the CD28 superagonist (Tegenero) drove strong cytokine secretion (and has shown extreme toxicity in early clinical studies back in 2006). Similar results were observed with EGFRxCD28. In addition, MUC16xCD28 was combined with MUC16xCD3, targeting two different epitopes of MUC16, to deliver “signal 1” and “signal 2”. The combination was safe, enhanced antitumor efficacy in ovarian cancer models, and is currently being tested in a Phase 1/2 clinical trial in patients with ovarian cancer.
CD8-targeted IL-2 drives potent anti-tumor efficacy and promotes action of tumor specific vaccines- Robert D. Schreiber, PhD – Washington University School of Medicine
Using his elegant T3 methylcholanthrene sarcoma cell line, which contains 3 dominant MHC class I (mutated [m]Lama4 and [m]Alg8) and class II (mutated [m]Itgb1) neoantigens and shows responsiveness to immune checkpoint blockade, Bob Schreiber delved into understanding the observation that delaying treatment with anti-PD-1 until day 8 lost effectiveness – an effect not seen with anti-CTLA-4. Inspired by the work of Zappasodi et al. (2018) showing the presence of a PD-1high class of non-Treg CD4+ T cells in patients who responded poorly to anti-PD-1 therapy, Schreiber and team used flow cytometry to reveal that in the T3 system, as tumors progressed, a similar phenotype was observed in mItgb1-specific CD4+ T cells. In parallel vaccination studies, optimization of peptide dose for the mItgb1 MHC class II peptide (always with a fixed dose of the MHC class I mAlg8 and mLama4 peptides) unexpectedly showed a bell shaped curve when tumor control was the readout (high-dose mItgb1 [1.5 ug; “HD”] being ineffective, and low-dose [0.0015 ug; “LD”] being highly effective). Vaccination with HD mItgb1 (and the MHC class I epitopes) also induced the same PD-1high class of non-Treg mItgb1-specific CD4+ T cells as the delayed treatment protocol. Moreover, vaccination with HD mItgb1 also abrogated the effectiveness of anti-PD-1 or anti-4-1BB, but again not anti-CTLA-4, even when given at day 3, as did adoptive transfer of T cells from HD-, but not LD-vaccinated mice. Single cell RNAseq of mItgb1-specific T cells revealed a cluster of mItgb1-specific CD4+ cells in HD-vaccinated mice that was distinct from Tregs or Tfh cells and was significantly increased in HD-vaccinated mice. Based on the observation that these cells did not produce IL-2, a cytokine critical for T cell help, Schreiber and his team reasoned that a targeted supply of IL-2 could overcome the generation of “helpless” CD8+ T cells. Using a cis-acting, CD8-targeted IL-2 mutein (IL-2 containing a mutation that eliminates binding to the IL-2 receptor alpha chain, but allows signaling through the beta and common gamma chains of the receptor, fused to a modified anti-CD8 anitbody), the researchers showed that this molecule restored the effectiveness of late anti-PD-1 therapy or of anti-PD-1 therapy given early in concert with HD vaccination. Interestingly, in the setting of HD vaccination, wild-type IL-2 was ineffective in restoring anti-PD-1 effectiveness.
Back to Top
By Ute Burkhardt, Ed Fritsch, Lauren Hitchings, and Maartje Wouters



