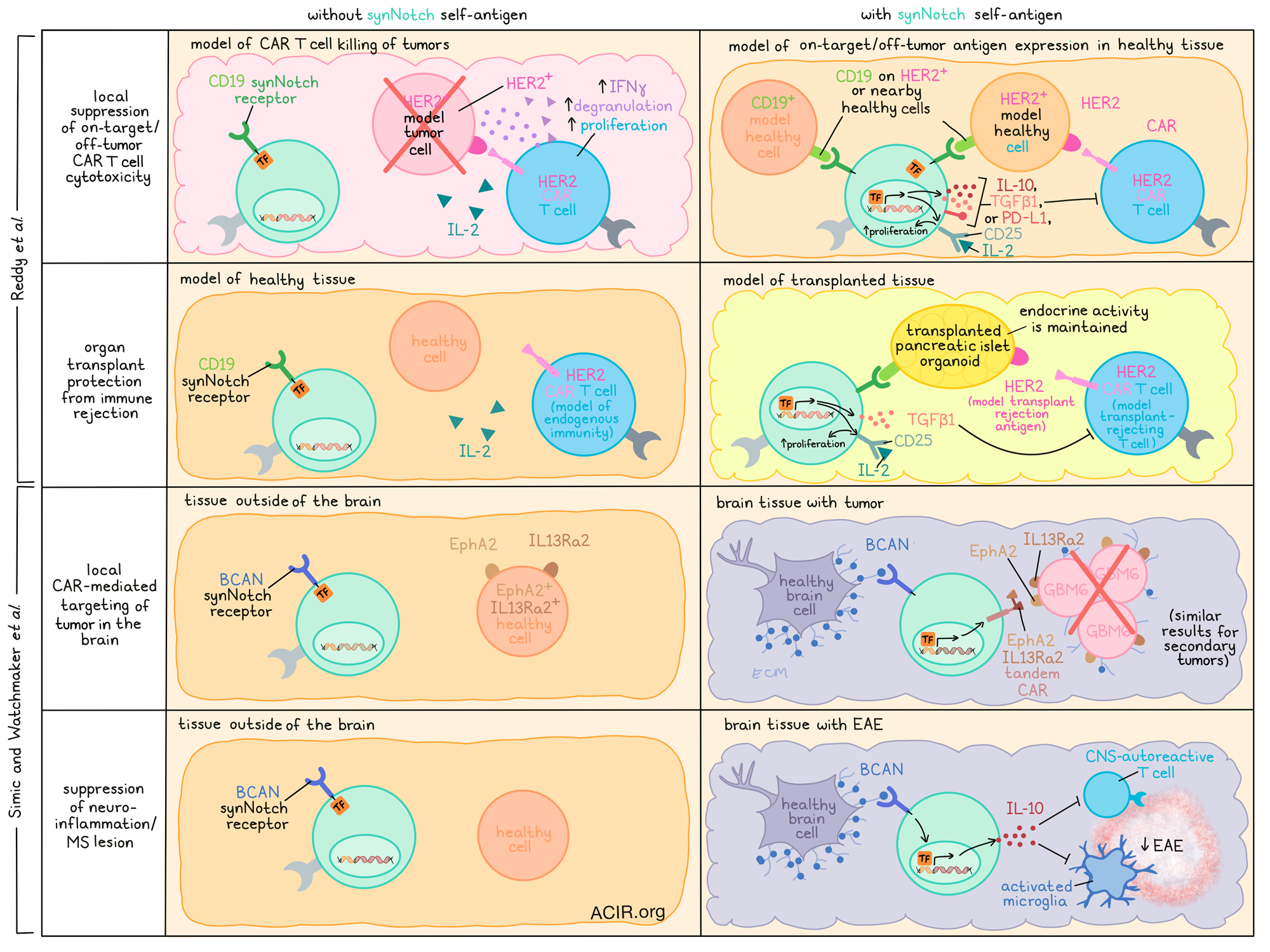
Immunotherapies are often used to target specific antigens or deliver specific payloads for the treatment of various diseases, but systemic toxicities or side effects in other tissues can limit the safety and efficacy of these treatments. Recently, two teams of researchers working with lead author Dr. Wendell Lim have demonstrated the use of synNotch-engineered CD4+ T cells directed towards tissue-specific antigens, triggering the local release of various disease-specific payloads. Reddy et al. utilized this technology to suppress on-target/off-tumor CAR T cell cytotoxicity and to suppress immune rejection in transplanted organs, while Simic and Watchmaker et al. demonstrated its ability to induce CAR expression only in the brain and to specifically deliver payloads that suppress neuroinflammation. Their results were recently published as back-to-back papers in Science.
SynNotch receptors consist of a variable extracellular recognition domain, a cleavable Notch-based transmembrane domain, and a transcriptional intracellular domain. Upon binding to its target antigen, the transmembrane domain of the synNotch receptor is cleaved, and the transcriptional domain is released, becoming free to enter the nucleus and activate expression of a transgene of choice from a synNotch-responsive promoter. Expression of the payload thus occurs only in the presence of the target antigen, and quickly decays in the absence of the target antigen, allowing for highly targeted local payload expression.
Utilizing synNotch technology, Reddy et al. aimed to develop synthetic suppressor cells by engineering CD4+ T cells with synNotch receptors that release immunosuppressive payloads to inhibit unwanted on-target/off-tumor cytotoxic activity related to CAR T cells. To show proof of principle, they designed synNotch receptors that, upon engagement with CD19, would release an immunosuppressive payload. In vitro investigations of various payloads and combinations of payloads showed that optimum payloads for inhibiting CAR T cell-mediated cytotoxicity included IL-10, TGFβ1, or PD-L1 (all immunosuppressive molecules) in combination with CD25, an IL-2 receptor subunit that, when expressed on the surface of the synNotch cells, acted as a cytokine sink for pro-inflammatory IL-2 and, as a secondary benefit, induced proliferation in the synNotch cells themselves.
Using CD19 as a model “off-tumor” antigen to trigger the synNotch circuit, the researchers showed that in cocultures of CD19+HER2+ cells and HER2-targeted CAR T cells, engineered CD19 SynNotch→TGFβ1+CD25 cells effectively inhibited the cytotoxic activity of CAR T cells, reducing IFNγ production, proliferation, and degranulation. Importantly, the synNotch cells did not inactivate themselves, as their signaling through synNotch was not inhibited by the suppressive payloads.
To test these synthetic suppressor cells in vivo, mice were transplanted with tumors on both flanks – tumors on both sides expressed HER2, but only one side expressed CD19 to activate the synNotch circuit. In these mice, the tumors expressing only HER2 were effectively eliminated by HER2-targeted CAR T cells, but those expressing both HER2 and CD19 were protected by CD19 synNotch→TGFβ1+CD25 cells, which accumulated in the CD19+ tumors and reduced the accumulation of the HER2 CAR T cells there.
Because synNotch cells release their payload and act through paracrine signaling, Reddy et al. found that synNotch cells also protected heterogenous mixtures of HER2+ cells and HER2+CD19+ cells from HER2 CAR T cell-mediated cytotoxicity in vitro, and protected tumors with as little as 25% CD19+HER2+ cells in mice. Together, these results suggest that synNotch can effectively be used to create a NOT gate, protecting tissues expressing selected healthy antigens from unwanted on-target off-tumor effects related to CAR T cells.
Reddy et al. also evaluated whether synthetic suppressor cells could be used to protect tissues from unwanted immune reactions in the case of transplantation. In vitro, pancreatic islet-like organoids of HER2+CD19+ beta cells could be protected against killing by HER2 CAR T cells with the addition of CD19 synNotch →TGFβ1+CD25 cells. The synNotch cells self-organized around attacking CAR T cells, preventing the formation of large clusters. In vivo, the same synNotch cells protected organoid transplants in 6 of 8 mice, with evidence of reduced CAR T cell proliferation and increased TGFβ1 accumulation in the transplant tissue, but not in the spleen or peripheral blood. Importantly, the protected transplants were functional and maintained endocrine activity.
Also investigating the potential of synNotch technology, Simic and Watchmaker et al. investigated whether synNotch cells could be used to deliver payloads specifically to the brain, as T cells can cross the blood-brain barrier. The team evaluated healthy CNS tissues for targetable antigens, and selected BCAN – an extracellular membrane protein – as a strong target antigen, due to its high expression levels in the brain, and the identification of an antibody that could be used to target it. In an effort to target glioblastoma, they then engineered a BCAN-targeted synNotch circuit that would induce the expression of a tandem CAR targeting either of two glioblastoma-associated antigens: ephrin type A receptor 2 (EphA2) and the IL-13 receptor a2 (IL13Ra2), which are not expressed in healthy brain tissue, but are expressed in some tissues outside of the brain. In vitro, BCAN synNotch→α-EphA2-IL13Ra2 CAR T cells were capable of killing of patient-derived GBM6 cells, which expressed both BCAN and the CAR targets, and this activity could be further enhanced with the addition of K562 cells, which expressed higher levels of BCAN and were unharmed by the synNotch cells.
In vivo, NCG mice implanted with GBM6 glioblastoma xenografts in the brain were treated with T cells bearing the BCAN synNotch→α-EphA2-IL13Ra2 CAR circuit. This resulted in increased tumor control, long-term remission, and increased survival compared to mice treated with control T cells. SynNotch cells expressing the CAR could be found throughout the brain, but cell killing was limited to tumors, where the CAR target antigens were expressed. SynNotch cells could also be found in other parts of the body, but did not express the CAR outside of the brain, in line with the absence of BCAN expression in other tissues. SynNotch cells also showed good persistence, and protected 3 of 4 mice from a rechallenge at day 86, with the fourth mouse showing slowed tumor progression. BCAN expression on tumor cells was not required for efficacy, as BCAN expression in nearby brain tissue was sufficient to activate expression of the CAR. However, tumors transplanted outside of the brain could not be controlled by synNotch T cells, as expression of the CAR rapidly decayed in the absence of BCAN.
Looking at whether synNotch cells could be used to clear secondary tumors, the researchers evaluated an BCAN synNotch→α-HER2 CAR circuit, and found that it could effectively clear HER2+ BT-474 tumor cells in the presence of BCAN+ K562 cells in vitro, and could eliminate HER2+ BT-474 tumors in the brains of mice. Similar results were observed with the TROP2-expressing TNBC tumor line BBT-20 and treatment using cells expressing a BCAN synNotch→TROP2 CAR circuit.
Next, Simic and Watchmaker et al. investigated whether synNotch cells could be used to target neuroinflammation and brain lesions associated with progressive MS. They developed a BCAN-targeted synNotch circuit that would induce secretion of IL-10 – a potent anti-inflammatory cytokine that, on its own, cannot cross the blood–brain barrier. Following successful in vitro testing against CNS-autoreactive T cells and activated microglia, the researchers found that delivery of BCAN synNotch→IL-10 cells to a murine model of experimental autoimmune encephalomyelitis (EAE) protected mice from severe disease, increasing their mobility and survival compared to untreated mice or mice treated with T cells that constitutively expressed IL-10. Treatment was well tolerated, with no accumulation of IL-10 in the periphery, no suppression of CD80 or CD86 on myeloid cells in the spleen, and no reduction of platelet counts, suggesting that the effects of IL-10 were limited to the CNS.
Overall, the results of these two studies show that synNotch-engineered CD4+ T cells can effectively deliver a variety of payloads to specific tissues, inducing highly localized effects that can be utilized in a number of disease settings. From suppressing unwanted off-tumor cytotoxicity, to inhibiting transplant rejection, to delivering and limiting treatments specifically to the brain, this technology allows for nested, multiscale targeting across a wide range of applications, and has the potential to make strides in cancer immunotherapy and beyond.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, Milo Simic, Payal Watchmaker, and Nishith Reddy answered our questions.

What was the most surprising finding of this study for you?
MS: Treatment of brain diseases is challenging and can be associated with off-target toxicity. That is because drugs can have multiple targets across the body. We built “smart cells” that recognize where they are in the body to specifically execute therapeutic actions only when they are in the brain. The “wow” moments were when we engineered those cells to kill cancer cells and tested them in a mouse model of glioblastoma, a fatal brain tumor: the tumors vanished! Amazingly, these “smart cells” could stay in the brain, even after clearing the tumor, and were ready to kill again if new cancer cells appeared. Excitingly, we were also able to use these “smart cells” to tackle neuroinflammation by delivering an anti-inflammatory factor!
PW: We were pleasantly surprised by the potency and persistence of our engineered T cells. Not only were they able to rapidly clear the brain tumors in the mice, but also they were long lasting and poised to strike back upon tumor recurrence. Time-lapse microscopy of brain-sensing T cell–astrocyte interactions showcased the effectiveness of our engineered sensor in turning on the cellular killing machinery. This visual evidence provided compelling insights into the robustness and the precision of our synthetic receptor.
NR: Current immunosuppressive drugs for treating inflammatory disorders completely shut down our immune systems, increasing susceptibility to cancer or infection. To address this challenge, we took inspiration from the way natural suppressor cells regulate our immune responses to engineer “synthetic suppressor T cells”, a highly programmable platform for locally targeted immune suppression. These cell therapies can treat inflammation in a specific targeted tissue, without producing systemic toxicities. In mouse models, we found these synthetic suppressor T cells could locally protect transplants from immune rejection or prevent cross-reactive toxicities of antitumor CAR T cells without systemic immunosuppression.
What is the outlook?
MS: It is a very exciting time as we embark on our journey to bring these “smart cells” to the clinic for the treatment of glioblastoma. This could, hopefully, expand the therapeutic options for these patients! Overall, what we built is a cell-based brain delivery platform that can be used to deliver user-defined therapies for a multitude of brain disorders.
PW: We are hoping to start a phase I clinical trial by the end of next year. This technology has the potential to be a game changer for otherwise deadly brain tumors.
NR: The programmability of synthetic suppressor T cells allows them to be retargeted to other tissues, such as the brain, as shown in Simic and Watchmaker et al., and tailored for specific inflammatory disorders, such as autoimmunity and transplant rejection. Importantly, we are no longer limited to native immune suppressor cell programs; we can now design customized cellular response programs that precisely execute a local therapeutic action. Additional studies will be needed to assess all translational considerations, but we hope these engineered cell therapies will benefit patients soon.
What was the coolest thing you’ve learned (about) recently outside of work?
MS: I have a Senegal parrot, and occasionally, he sneezes. I recently learned that, unlike us, they sneeze dry!
PW: I recently had the opportunity to attend Kristin Hege’s talk about her thru-hiking experience of the Pacific Crest Trail. “There is always another hill to climb” resonates with both CAR T cell research and the PCT.
NR: I recently visited the Great Barrier Reef and was in awe at all the incredible natural biodiversity. It is remarkable to think that evolution has generated such complex biological phenomena, from the intricate behaviors of coral reefs to the complex interactions of our immune systems. There is still so much we don’t know about biology!



