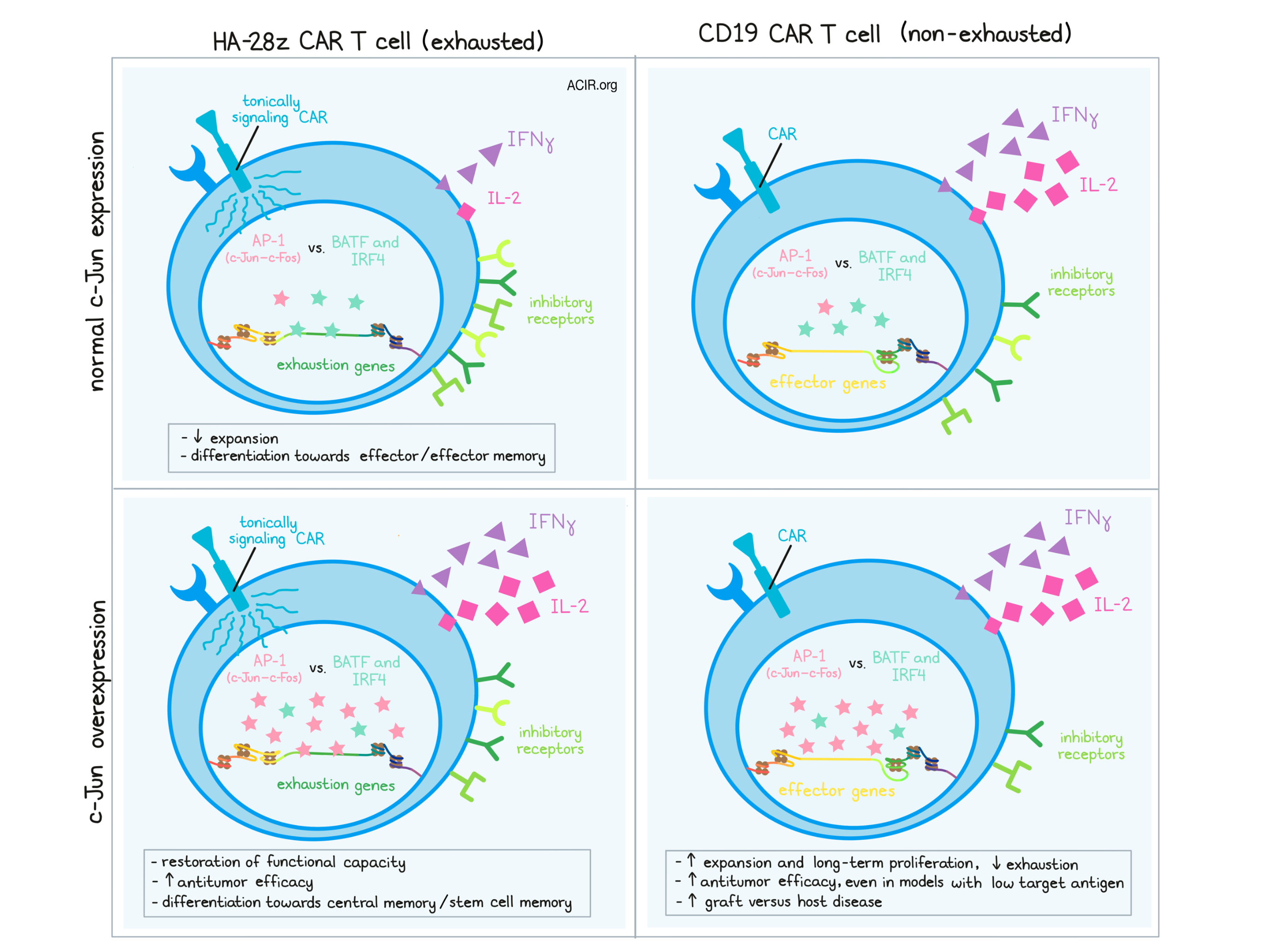
In an effort to understand and overcome exhaustion in adoptively transferred CAR T cells, Lynn et al. developed a cellular model for exhaustion and interrogated its programming. Their results, recently published in Nature, reveal a critical role for the transcription factor component c-Jun (part of the canonical AP-1 c-Fos–c-Jun heterodimer) in preventing exhaustion. Capitalizing on this information, the researchers also show that c-Jun overexpression can be used to enhance the efficacy of CAR T cell therapy in several models.
To first model T cell exhaustion, Lynn et al. incorporated a tonically signaling HA-28z CAR into T cells to induce hallmark features of exhaustion, including reduced expansion, increased expression of inhibitory molecules, increased differentiation towards effector phenotypes, reduced IFNγ production, and markedly reduced IL-2 production following stimulation compared to control CD19-28z CAR T cells. Differential gene expression and RNAseq data validated this exhausted state.
To probe the molecular mechanisms driving differential gene expression in tonically signaling HA-28z CAR T cells, the researchers performed ATACseq analysis, which revealed extensive differential chromatin accessibility driven primarily by the expression of the HA-28z CAR. Clustering the top 5000 differentially accessible regions showed evidence of a consistent epigenetic state across HA-28z-expressing cells, with a consistent pattern among CD4+, CD8+, naive, and memory subsets. This epigenetic state showed increased chromatin access to genes like CTLA4 and reduced chromatin access to memory-associated genes like IL7R, consistent with exhaustion.
Analyzing the transcriptional programs associated with the observed exhaustion-associated epigenetic state, Lynn et al. found that AP-1–bZIP and bZIP–IRF binding motifs were particularly accessible in exhausted CAR T cells, as were NF-κB, NFAT, and RUNX transcription factor motifs (in some clusters). Paired RNAseq analysis revealed increased bZIP and IRF transcription factor expression (most notably JUNB, FOSL1, BATF, BATF3, ATF3, ATF4, and IRF4), and protein analysis confirmed the increase in JunB, IRF4, and BATF3. Importantly, the researchers noted that BATF and IRF4 – transcription factors that infamously drive regulatory programs – were present at higher relative levels than c-Jun, which dimerizes with c-Fos to form the canonical AP-1 transcription factor that drives IL-2 expression. As AP-1, BATF, and IRF4 are known to compete for access to binding motifs, Lynn et al. hypothesized that an imbalance between these activating and regulatory transcription factors may underlie exhaustion programming.
To test this hypothesis, Lynn et al. engineered tonically signaling HA-28z CAR T cells to overexpress AP-1. This effectively restored cellular production of IL-2, and was dependent on c-Jun, but not c-Fos. HA-28z CAR T cells engineered to overexpress just c-Jun showed similar restoration of IL-2 and IFNγ production; to fully restore IL-2 production, c-Jun overexpression was required during both T cell expansion and antigen stimulation. JUN-HA-28z CAR T cells also showed increased functional capacity and a shift from effector and effector memory differentiation towards stem cell memory and central memory differentiation.
In non-tonically signaling CD19-specific CAR T cells, c-Jun overexpression did not affect IL-2 or IFNγ production, but did enhance long-term proliferative capacity and reduce expression of exhaustion markers in vitro. In vivo, c-Jun overexpression in transferred CAR T cells enhanced their capacity for expansion. In one model, however, this also accelerated the progression of graft-versus-host disease.
Based on their evidence that c-Jun overexpression prevents the induction of exhaustion programs, the researchers looked at ATACseq and RNAseq data to compare JUN-HA-28z CAR T cells to HA-28z CAR T cells. While c-Jun overexpression did not appear to change the epigenetic profile within cells, it did substantially alter the transcriptome, reducing expression of exhaustion-associated genes and increasing expression of memory-associated genes. Unsurprisingly, many of the genes affected by c-Jun overexpression were found to be enriched for AP-1 family binding sites.
Lynn et al. then hypothesized that c-Jun overexpression may prevent exhaustion directly, by activating AP-1 target genes, and/or indirectly, by disrupting the activity of immunoregulatory transcriptional complexes. To test which of these mechanisms might be at play, the researchers evaluated a panel of c-Jun mutants predicted to be deficient in transcriptional activation, DNA binding, or dimerization. Two mutants deficient in transcriptional activation restored IL-2 production as effectively as wild-type c-Jun, while a third demonstrated partial rescue of IL-2. Mutants deficient in DNA binding or dimerization, on the other hand, did not rescue cytokine production in exhausted HA-28z CAR T cells. Overexpression of c-Jun decreased levels of exhaustion-driving AP1–IRF and JunB–BATF transcriptional complexes, as well as total chromatin-bound JunB, BATF, and BATF3. Further, knocking out JunB, BATF, or especially IRF4 increased production of IL-2 and IFNγ. Together, these results support the notion that c-Jun overexpression staves off exhaustion programming primarily by mediating the displacement of immunoregulatory transcription factors.
Finally, to test antitumor efficacy, c-Jun-overexpressing CAR T cells were adoptively transferred into various murine tumor models. Compared to HA-28z CAR T cells, JUN-HA-28z CAR T cells showed enhanced antitumor activity in a Nalm6-G2D+ leukemia model. Importantly, overexpression of c-Jun in CD22-specific or CD19-specific CAR T cells enhanced antitumor efficacy in leukemia models with low target antigen expression. In a solid tumor setting, JUN-Her2-BBz CAR T cells prevented the growth of 143B osteosarcoma tumors and improved long-term survival of mice. The JUN-Her2-BBz CAR T cells showed greater expansion, greater accumulation in tumors, reduced expression of exhaustion markers, longer-term persistence, and enhanced cytokine production upon restimulation ex vivo compared to control Her2-BBz CAR T cells. A small cluster of JUN-Her2-BBz CAR T cells characterized by high IL7R expression persisted long-term in mice, consistent with the maintenance of a self-renewing memory-like population. Similar results were observed when treating 143B sarcoma with JUN-G2D-BBz CAR T cells.
Overall, Lynn et al. show that a functional deficiency in c-Jun mediates dysfunction in exhausted T cells, and that forcing overexpression of c-Jun can prevent exhaustion programming by outcompeting exhaustion-inducing immunoregulatory transcription factors. This strategy could be used to prevent exhaustion and enhance the efficacy of CAR T cells.
by Lauren Hitchings



