Given that antibodies to 4-1BB (a member of the tumor necrosis factor receptor superfamily that is expressed on activated T cells and other activated immune cells) have demonstrated antitumor effects in preclinical cancer models, Buchan, Dou, and Remer et al. set out to delineate the ways in which these antibodies promote antitumor immunity, and how the specific mechanisms depend on the antibodies’ Fab and Fc regions. The results were recently published in Immunity.
The researchers began by generating murine mIgG1 and mIgG2a versions of a anti-4-1BB monoclonal antibody (mAb). When tested in three solid tumor models (CT26 colon carcinoma, EG7 thymoma, NXS2 neuroblastoma), the mIgG2a antibody resulted in 80% long-term survival while the mIgG1 antibody did not produce much long-term benefit.
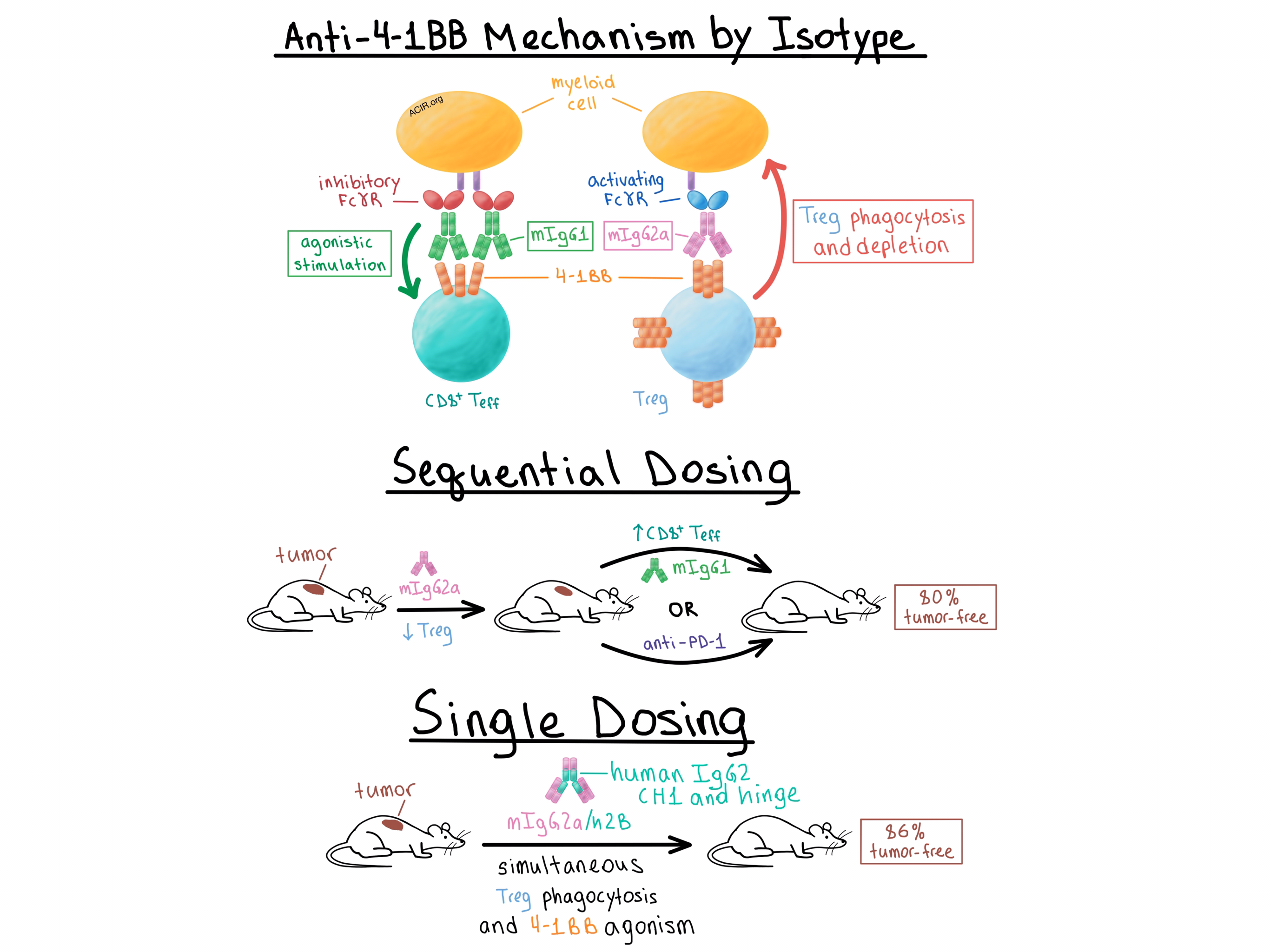
Buchan, Dou, and Remer et al. sought to understand the mechanistic differences between the two antibody isotypes that might explain their differential efficacy. Analysis of the antibodies’ binding to activating and inhibitory Fcγ receptors (FcγRs) showed that, as expected, mIgG1 was more likely to bind inhibitory FcγRs and mIgG2a was more likely to bind activating FcγRs, while both isotypes bound 4-1BB equally. Aware that 4-1BB is upregulated at much higher levels on intratumoral Tregs than on effector T cells in murine and human solid cancers, the researchers looked into Treg depletion.
The team found that mIgG2a, but not mIgG1, efficiently reduced the percentage of intratumoral Tregs in CT26 tumor-bearing wild-type mice. However, in mice that lacked the inhibitory FcγRIIB, mIgG1 also became quite effective at depleting intratumoral Tregs. In order to confirm these results, the researchers utilized bone marrow-derived macrophages from wild-type mice and showed that mIgG2a induced phagocytosis of 4-1BB-expressing target T cells, while mIgG1 did not. A separate experiment found that the efficiency of target cell depletion was directly correlated with the level of 4-1BB expression. Furthermore, in EG7 tumor-bearing mice with a selective knockout of 4-1BB only on Tregs, mIgG2a lost its Treg-depleting activity and long-term survival benefit. Together, these experiments demonstrated that Treg depletion depended on the binding of the antibody to both the 4-1BB on the target cell and an FcγR, presumably on a myeloid cell.
Diving deeper into the role of FcγRs in the therapeutic effect of anti-4-1BB mAbs, the researchers noted that while mIgG2a appeared to rely on activating FcγRs for its depleting functionality, it retained its antitumor effect in mice lacking activating FcγRs. In addition, mIgG1 improved its antitumor effect in the absence of activating FcγRs. When all FcγRs were absent, neither isotype provided any therapeutic effect. Together, these results suggested that the FcγRs compete for binding of both antibody isotypes, and in the absence of activating FcγRs, both isotypes successfully bind the inhibitory FcγRIIB and deliver the costimulatory signal.
Next, Buchan, Dou, and Remer et al. explored how isotype affects agonist activity. In vitro and in vivo, mIgG1 was more effective than mIgG2a in stimulating T cells. This effect was not dependent on the presence of Tregs, indicating that mIgG1 directly targets 4-1BB on effector T cells. When the two antibody isotypes were mixed, the costimulatory effect of mIgG1 disappeared, demonstrating that the two isotypes equivalently compete for binding of 4-1BB on T cells. Mechanistically, knockout experiments revealed that the agonistic activity of mIgG1 was dependent on the inhibitory FcγRIIB. Furthermore, mIgG2a demonstrated agonistic activity in the absence of activating FcγRs, suggesting that it bound the inhibitory FcγRIIB in these circumstances. In tumor-bearing mice, both isotypes led to an increase in proliferation and activation of tumor-infiltrating CD8+ T cells, although the magnitude of this effect was greater with mIgG1. Overall, these experiments showed that the therapeutic efficacy of mAbs was dependent on the mAb Fc binding profile and the availability of particular FcγRs. Specifically, mIgG1 exerted its effect primarily by directly activating CD8+ T cells following cross-linking via the inhibitory FcγRIIB, while mIgG2a preferentially bound activating FcγRs and promoted antitumor immunity primarily by depleting Tregs.
The team then considered that the differential mechanisms of action of the two isotypes could be combined to maximize the antitumor effect. While concurrent administration of mIgG1 and mIgG2a led to reduced antitumor efficacy compared with mIgG2a alone, sequential administration of mIgG2a first to deplete Tregs, followed by mIgG1 to stimulate effector T cells, reduced tumor size and increased the percentage of tumor-free mice to 80%. Sequentially dosing mice with mIgG2a followed by anti-PD-1 similarly increased the long-term response rate to 80%, compared with 20-25% response with either monotherapy. Meanwhile, combining mIgG1 with anti-PD-1 did not improve the response rate, once again demonstrating the importance of understanding the effect of isotype.
Finally, the researchers created a single anti-4-1BB antibody capable of simultaneously depleting Tregs and stimulating effector T cells by substituting a modified human IgG2 CH1 and hinge regions for the equivalent domains in the murine mIgG2a constant region – a change known to enhance FcγR-independent agonistic properties for other TNFR family members. The resulting anti-4-1BB mIgG2a/h2B antibody was more agonistic in vitro than the mIgG2a isotype while retaining its phagocytosis-inducing properties. In vivo, monotherapy with the antibody depleted intratumoral Tregs, stimulated tumor-infiltrating CD8+ T cells, and led to cure of 86% of EG7 tumor-bearing mice compared with 53% cure rate with mIgG2a. Thus, the team demonstrated that a single mAb could be rationally engineered, based on an understanding of the contributions of Fc binding to FcγR, to effect both depletion and agonism in order to improve the antitumor response of mAbs targeting 4-1BB and potentially other targets.
by Anna Scherer



