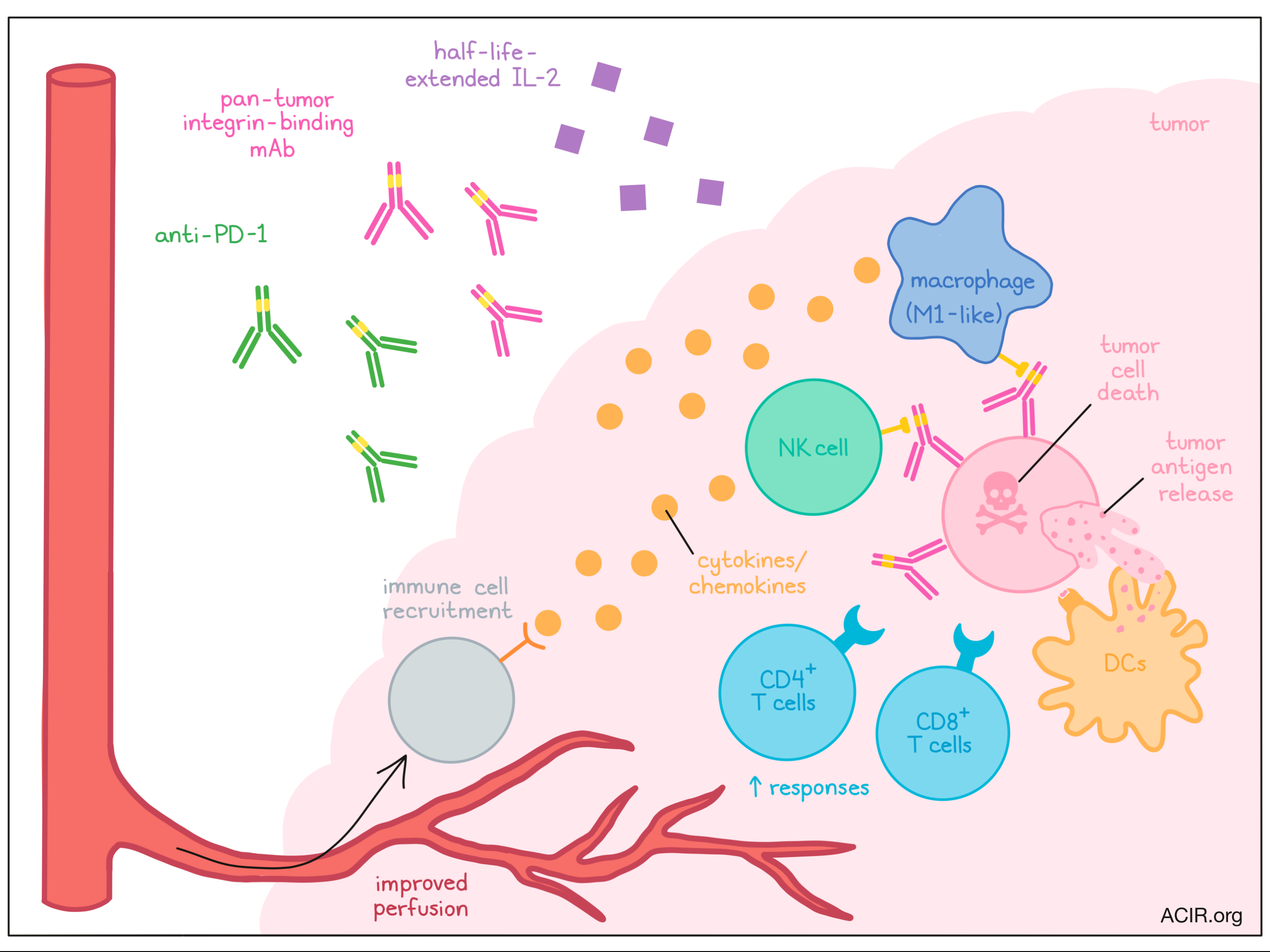
While the precise features enabling patient response to immune checkpoint blockade (ICB) remain unclear, “hot” tumors displaying immune activity and inflammation tend to respond better than quiet, “cold” tumors. Therapies that flip this switch could extend ICB response to a broader set of patients. In Cell Reports, Wang et al. reported on a three-pronged immunotherapy strategy to activate the tumor microenvironment (TME) and boost ICB efficacy, revealing a critical role for innate immunity.
Previously, these researchers developed a four-arm therapy (called “AIPV”) consisting of: (A) an antitumor mAb, (I) extended serum half-life IL-2, (P) anti-PD-1, and (V) a peptide vaccine. They found that a multi-dose AIPV treatment schedule excited the TME and regressed large tumors. In this current work, Wang et al. developed this strategy to facilitate clinical translation. The authors began by tackling two key features: the extended dosing regimen and patient specificity.
First, the researchers tested whether a single dose of AIPV could sufficiently inflame the TME to respond to subsequent ICB (anti-PD-1 + anti-CTLA-4). Melanoma-bearing mice were treated with AIPV consisting of an anti-TYRP-1 antibody, IL-2, anti-PD-1, and TRP2 peptide. Within one day, the single dose increased pro-inflammatory cytokine levels (e.g., IFNγ, TNFα) within the TME. Importantly, while ICB alone had no response in this tumor model, ICB following the single dose of AIPV (called AIPV + ICB) elicited tumor regressions, comparable to multiple doses of AIPV.
Next, Wang et al. sought to generalize this strategy, replacing anti-TYRP-1 with an antibody capable of binding integrins overexpressed in a variety of cancer cell lines. AIPV + ICB with this antibody was also effective, and in fact, was equally effective with the removal of the vaccine component (note: the AIV + ICB strategy was also equally effective, but required a tumor target-specific vaccine). This new combination (AIP + ICB), now broadly generalizable, led to tumor regressions in the B16F10, TC-1, and KP models, as well as in a tamoxifen-inducible BrafV600E/Ptenfl/fl melanoma model.
The researchers next characterized which cell types mediate the response to AIP therapy. Depleting CD4+ or CD8+ T cells, or applying AIP to Batf3-/- mice lacking cDC1s, reduced therapeutic efficacy, indicating the importance of each of these cell types in the response. Within the TME, AIP increased tumor cell apoptosis, calreticulin expression, and tumor antigen uptake by CD103+ DCs; expression of costimulatory and homing molecules, CD86 and CCR7, was increased on these DCs. To determine whether these DCs could prime CD8+ T cells, pmel (melanoma gp100-specific) T cells were adoptively transferred prior to AIP therapy. AIP treatment substantially increased pmel proliferation just one day after AIP treatment, altogether suggesting that AIP can rapidly release tumor antigen for uptake by DCs, and priming of CD8+ T cell responses.
Wang et al. next considered how AIP might induce tumor cell death, a key piece in this process. They hypothesized that innate immune cells might interact with the anti-integrin antibody, and to test this, depleted several innate cell types. They found that loss of NK cells (anti-NK1.1) or macrophages (anti-F4/80), but not neutrophils (anti-Gr-1), reduced tumor cell death and efficacy after AIP + ICB. Interestingly, modifying the anti-integrin antibody to prevent Fcγ receptor binding also decreased AIP-induced tumor cell death and therapeutic response, suggesting that NK cells or macrophages may engage with the antibody through Fc receptors for tumor cell killing (possibly through ADCC). Consistent with this hypothesis, AIP + ICB was ineffective in Fcgr3-/- mice (lacking FcγRIII), but could be rescued by adoptive transfer of wild-type NK cells or macrophages. Thus, NK cells and macrophages are key early AIP responders, engaging the antibody through Fcγ receptors to induce cancer cell death.
Given the important role of NK cells and macrophages in the AIP response, the team next interrogated gene expression changes in these cell types. Bulk RNAseq found that AIP broadly activated NK cells and macrophages, which upregulated IFN-related and effector genes (e.g., Gzmb, Ifng), compared to cells from untreated tumors. In addition, NK cells in AIP-treated tumors showed features of memory and metabolic activity, while macrophages expressed an M1-like phenotype. All three AIP components (versus only AI, IP, or AP) drove these changes most robustly. Notably, the largest difference in gene expression was found between AIP and IP, further highlighting the role of the antibody in engaging NK cells and macrophages.
In their analysis, Wang et al. also found gene expression changes in NK cells and macrophages relating to chemokine secretion and tumor vasculature – both important for immune cell recruitment. One day after AIP, a variety of chemokines including CXCL9, CXCL10, and FLT3L increased within tumors, and over the following two days, an influx of DCs, NK cells, macrophages, and T cells was observed. Interestingly, depleting NK cells or macrophages reduced cell recruitment.
Next, the researchers investigated the tumor vasculature. AIP-treated tumors increased patent blood vessels (which expressed adhesion molecules), perfusion of an i.v.-injected dye, and pericyte coverage, and reduced intratumoral hypoxia compared to tumors left untreated or treated with ICB alone. Again, depletion of NK cells or macrophages abolished these beneficial changes. Intriguingly, blocking IFNγ or TNFα also prevented these changes, recapitulated in mice containing Ifng-/- (but not wild-type) NK cells or macrophages. These results suggested that NK cells and macrophages, by secreting inflammatory cytokines and chemokines and supporting blood vessel normalization, can recruit further immune cells and develop a “hot” TME.
In summary, Wang et al. developed a multi-immunotherapy strategy to heat up a cold TME, activating NK cells and macrophages to kill cancer cells, recruit additional effectors, and remodel the tumor vasculature. Altogether, this therapy improved the subsequent application of ICB in otherwise refractory tumor models. As immunotherapies continue to develop, understanding how they may synergize with one another may enable more effective and broadly applicable cancer treatments.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, first author Chensu Wang answered our questions.

What prompted you to tackle this research question?
We were intrigued by the limited efficacy of immune checkpoint blockade (ICB) in patients with immunologically “cold” compared to “hot” tumors. We wanted to find out an efficacious treatment that converts “cold” tumors into “hot” ones, and thus synergizes with ICB. A follow-up study on our previous work – a potent four-component immunotherapy (termed AIPV, Moynihan et al. 2016) – revealed striking changes induced in the tumor microenvironment within a week following a single dose, which was likely due to responses from innate immune cells. This led us to hypothesize that a simplified regimen based on AIPV treatment could activate the innate immune system and prime tumors to respond to treatment with ICB alone.
What was the most surprising finding of this study for you?
The most surprising finding of this study for me was how tumor vascular structures could be quickly remodeled following the activation of NK cells and macrophages, both in terms of the structures of blood vessels and the coverage and integrity of perivascular coverage. This suggests that NK cells and macrophages are able to quickly engage a more comprehensive and complex signaling network besides direct killing of tumor cells and recruitment of other immune cells. Further studies are needed to better understand how they communicate with vascular endothelial cells, fibroblasts, pericytes, and smooth muscle cells to facilitate the extravasation of T cells into the tumor site.
What was the coolest thing you have learned (about) recently outside of work?
I recently learned how to differentiate sea lions from seals, as I became a more regular visitor to the pinniped hangouts on the shore after moving to California. Sea lions have external ear flaps and seals don’t, which one can easily tell when watching them from a distance. Sea lions also bark very loudly (usually a male), while seals are only capable of low grunts.



