CD8+ T cells are often the focus of research in cancer immunotherapy, but recent evidence has suggested that CD4+ T helper cells (particularly T follicular helper [TFH] cells) and B cells (particularly germinal center [GC] B cells) may also play important behind-the-scenes roles in supporting CD8+ T cells and antitumor immune responses. Cui et al. recently investigated the roles of these cells in the setting of lung adenocarcinoma (LUAD), and their results were published in Cell.
To begin, Cui et al. analyzed scRNAseq data from TCGA and other published datasets and found that B cells and CD4+ T cells were among the most abundant immune cell types present in LUAD tumor samples. One algorithm suggested that a large proportion of CD4+ T cells were TFH cells, and additional analyses of cells confirmed clusters of TFH cells, which expressed TFH-associated genes and the TFH signature cytokine IL-21. Clustering analysis also showed evidence of GC B cells. Both the TFH and GC B cells were enriched in tumors compared to healthy lung tissues, and gene signatures for each of these cell types correlated with prolonged overall survival. Further, these cell types correlated with one another (suggestive of a relationship between them within tumors) and with Th1 and CD8+ effector T cell signatures.
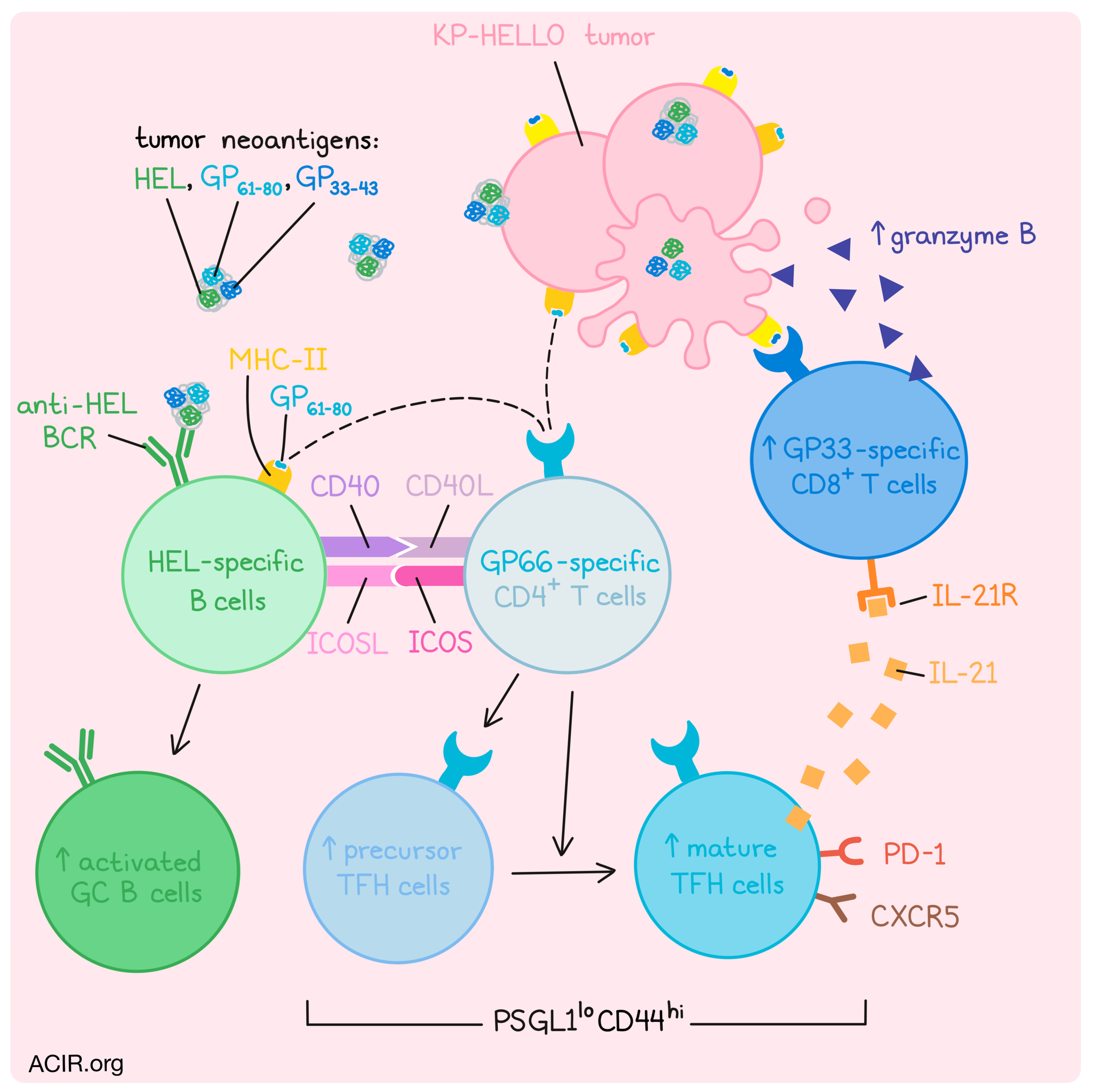
To better study these cell types and the interactions between them, Cui et al. first developed an experimental mouse model. To begin, they implanted a LUAD cell line derived from KP mice and confirmed the development of precursor (CD44hiPSGL1lo) and mature (CD44hiPSGL1loCXCR5+PD-1+) TFH cells. Next, they used their previously developed NINJA system to program KP lung tumor cells to express two model LCMV antigens as tumor neoantigens. The first neoantigen, GP33-43, is recognized by a GP33-specific CD8+ TCR-transgenic T cells, while the second neoantigen, GP61-80, is recognized by GP66-specific CD4+ TCR transgenic T cells. Building on the NINJA system, the researchers also developed the HELLO system, which incorporated programming of KP lung tumor cells for expression of heg-egg lysozyme (HEL), a model foreign B cell antigen recognized by both MD4 and SWHEL B Cell Receptor (BCR)-transgenic B cells, linked on the same secreted polypeptide as the GP33-43 and GP61-80 epitopes.
In vitro, KP-HELLO tumor cells effectively presented the HEL antigen and activated HEL-specific B cells. Further, activated HEL-specific B cells were exclusively able to present the GP61-80 antigen (delivered to these B cells as the secreted HEL–GP33-43–GP61-80 polypeptide) to GP66-specific CD4+ T cells. While these cells were also capable of presenting epitopes on MHC-I, they were not able to present the GP33-43 antigen to GP33-specific CD8+ T cells, likely due to an inability to process that particular model antigen.
Moving into mice, the researchers showed that implanted KP-HELLO tumors elicited the development of neoantigen-specific precursor and mature TFH cells. To investigate the specific contribution of the B cell-recognized neoantigen, the researchers compared KP-HELLO tumors against KP-NINJA tumors, which did not express HEL. Compared to the KP-NINJA, the KP-HELLO tumors showed a greater increase in IgDlo-activated B cells, many of which were consistent with GC B cell phenotype, as well as a greater increase in precursor and mature TFH cells, providing evidence for the hypothesis that tumor-specific B cells support the development of tumor-specific TFH cells.
Next, looking at the extent of immune control in these models, the researchers found that compared to in wild-type mice, KP-HELLO tumors grew faster in immunodeficient mice, and in mice lacking B cells, CD4+ T cells, or just TFH CD4+ T cells. Mice lacking B cells showed reduced development of precursor TFH cells and fully impaired development of mature TFH cells. Tumor control could be rescued with the adoptive transfer of HEL-specific B cells, but not non-specific B cells. Impaired development of TFH cells was also observed in mice bearing KP-NINJA tumors, suggesting that tumor-specific B cell responses are essential to the development of TFH cells. This suggested that B cells control the development of CD4+ T cell compartment by promoting TFH differentiation. Tumor control could be also restored in TFH cell-deficient mice. Adoptive transfer of tumor-specific CD4+ T cells, a portion of which differentiated into TFH cells, restored tumor control and polyclonal GC B cell responses, suggesting bidirectional interactions between B cells and CD4+ T cells. In line with this, interactions between CD40/CD40L and ICOS/ICOSL were required for mature TFH and GC B cell responses and for tumor control.
Turning their attention towards how GC B and TFH cells might relate to CD8+ T cells, Cui et al. first showed through antibody-mediated depletion that CD8+ T cells played a key role in control of KP-HELLO tumors in wild-type mice. Looking at CD8+ T cell phenotypes, they found that about 40% of CD8+ T cells displayed an effector phenotype, expressing granzyme B and PD-1. Meanwhile, in TFH or B cell-deficient mice, effector CD8+ T cell populations in tumors were reduced. Interestingly, in KP-NINJA models, B cell deficiency had no impact on effector CD8+ T cell populations, suggesting that the incorporation of the B cell-recognized neoantigen in the KP-HELLO model drove effector CD8+ T cell functions via a B cell- and TFH cell-dependent mechanism. Similar observations were made in an autochthonous mouse model in which tumors developed in the lungs.
Investigating the relationships between immune cells, the researchers turned their attention towards the signature TF cytokine IL-21, which has previously been implicated as a driver of effector T cell functions. Using their models, they showed that CD4+ cells, mainly TFH and some TFH precursor cells, did express IL-21 in tumors, and that this was strongly dependent on the presence of both B cells and B cell-recognized neoantigens. Further, the IL-21 receptor was expressed on CD8+ effector T cells in the tumor and dLN. Blocking this receptor increased the rate of tumor progression and strongly reduced effector CD8+ T cells among TILs, suggesting that B cell-dependent and TFH cell-produced IL-21 acts on CD8+ effector T cell to induce robust antitumor responses.
Together these results suggest that presence of B cell and CD4+ T cell epitopes drives interactions between tumor-specific B cells and tumor-specific CD4+ T cells, which provide bidirectional signals that support the activation and differentiation of GC B cells and the differentiation of CD4+ T cells into precursor and mature TFH cells. Mature TFH cells, in turn, produce IL-21, which supports antitumor CD8+ T cell responses. This research enhances the current understanding of the mechanisms that support immune responses in the tumor microenvironment and relevant lymph nodes, and provides an entry point for potential therapeutic manipulation.
Write-up and image by Lauren Hitchings



