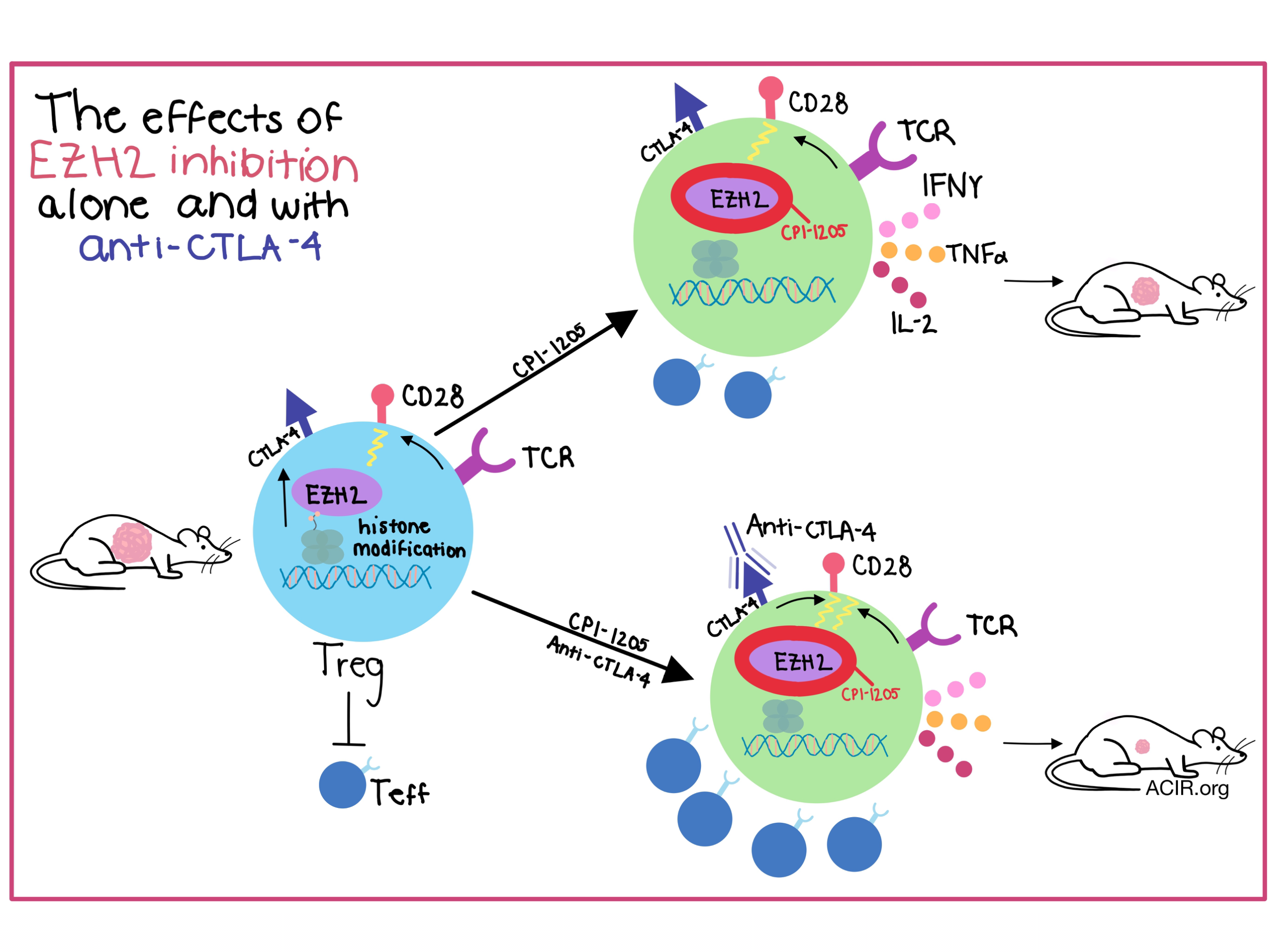
Regulatory T cells (Tregs) are frequently found in high quantities in cancers, where a high load of Tregs corresponds to a poor prognosis, but systemic depletion of Tregs can unleash severe autoimmunity. In the search for new ways to safely deplete Treg populations, Wang et al. in their paper published in Cell and Goswami et al. in the Journal of Clinical Investigation uncovered a new target induced in activated Tregs, enhancer of zeste homolog 2 (EZH2), an enzyme involved in histone methylation and transcriptional repression. Small molecule inhibition of EZH2 not only reduced the net suppressive activity of tumor Tregs, but also re-programmed Tregs into effector-like cells, enhancing antitumor effects.
Wang et al. began their experiments by exploring the effects on immune cells of EZH2 inhibition with CPI-1205, a drug being tested clinically for direct antitumor effects to modulate cancer progression. In immunocompetent mice implanted with MC38 murine colorectal cancer, EZH2 inhibition caused a dramatic inhibition of tumor progression, but in T cell-deficient Rag-1-/- mice, tumor growth was accelerated, indicating that EZH2 inhibition involves T cells. An increased portion of CD8+ T cells in tumors further confirmed these findings. Tumor-infiltrating Tregs (TI-Tregs) that had been treated with CPI-1205 exhibited significantly reduced levels of trimethylated histone H3 as a result of the inhibition of EZH2’s enzymatic activity in Tregs. While TI-Treg populations weren’t decreased by EZH2 inhibition, the ratio of CD8+ T cells to TI-Tregs increased. These effects were targeted to TI-Tregs as EZH2 protein and its associated histone modifications were not observed in Teff in tumors or in Tregs in tumor-draining lymph nodes. Translational studies with human melanoma, colorectal, non-small cell lung, and breast cancers confirmed that TI-Tregs expressed higher levels of EZH2 protein or RNA in comparison to Teff or non-tumor Tregs.
Turning to genetic approaches to confirm their findings, Wang et al. found that mice that had Ezh2 constitutively deleted in Tregs completely rejected MC38 and prostate carcinomas after an initial tumor growth phase. A poorly immunogenic B16F10 model showed only growth reduction in the Treg EZH2 knock-outs, but coupling with vaccination led to complete tumor rejection.
Insertion of a cre-activatable red-fluorescent protein marker allowed the team to follow the fate of Foxp3-expressing cells after EZH2 deletion. Genetic deletion of Ezh2 greatly reduced Foxp3 expression, lowering the number of TI-Tregs that were FOXP3+, and shifting the T cell population in tumors towards CD8+ cells rather than Tregs. Importantly, the remaining FOXP3+ Tregs shifted from secretion of the immunosuppressive cytokine IL-10 toward a proinflammatory cytokine producing state (TNFα, IFNγ, IL-2) in tumors. This enhanced polyfunctionality was confirmed by the restimulation of TI-T cells in vitro. Overall, the results of the experiments by Wang et al. indicated that Ezh2-deficient TI-Tregs had an altered, pro-inflammatory state that helped elicit stronger tumor immunity. Wang et al. hypothesized that because EZH2 is CD28-dependent for induction, EZH2 inhibition would be well-combined with immune checkpoint blockades that also target CD28, such as anti-CTLA-4.
The combination of EZH2 inhibition and anti-CTLA-4 was specifically explored in the work by Goswami et al. Their experiments began, similarly to Wang et al., by knocking out Ezh2 in Foxp3creEZH2fl/fl C57BL/6 mice. Following inoculation with a bladder cancer cell line (MB49) they observed that the knockout mice had significantly less tumor growth when compared to control mice. The loss of EZH2 function resulted in increased CD8+ activated Teff cells and, as also observed by Wang et al., the team noted a change in phenotype of the Foxp3+ cells toward effector-like T cells, enhancing antitumor immunity. Comparing different drugs to knock out EZH2 function, CPI-1205 was found to have the strongest effect on suppressing the differentiation of murine inducible Tregs (iTregs). Proinflammatory pathways, including Th1 and Th2, were upregulated in iTregs after treatment with CPI-1205, and RNA sequencing and analysis of secreted cytokines demonstrated that CPI-1205 inhibition also skewed the Treg cells toward a proinflammatory phenotype. EZH2 inhibition also decreased expression of genes required for Treg stability, including Nrp1 and Bach2; deficiencies in genes like Nrp1 in Tregs coupled with IFNγ production have been tied to instability in neighboring Tregs, which further restores antitumor immunity. Aiming to confirm their results in a human system, Goswami et al. showed that human iTregs treated with CPI-1205 reduced Foxp3 expression in a dose-dependent fashion and were found to lose their suppressive abilities, resulting in an increase in the proliferation of conventional T cells.
Pulling together the observations that activation of CD28 signaling causes EZH2 expression and that CD28 signaling was enhanced by anti-CTLA-4 therapy, Goswami et al. hypothesized that anti-CTLA-4 therapy might enhance EZH2 expression, potentially subverting the anti-CTLA-4 antitumor effect. The link between increased EZH2 expression and anti-CTLA-4 therapy was confirmed when human CD4+ Teff, Tregs, and CD8+ T cells were tested at baseline and post-ipilimumab treatment, and an increase in EZH2 levels in all three types of T cells after ipilimumab treatment was observed. Because of this link, Goswami et al. tested anti-CTLA-4 and CPI-1205 in combination therapy, and found, as hypothesized, that combination therapy significantly reduced tumor growth and increased survival in the MB49 and B16F10 models.
Wang et al. demonstrated that CPI-1205 mediated EZH2 inhibition, altered TI-Tregs toward a proinflammatory state, and increased the presence of effector T cells, and Goswami et al. used the relationship between CD28 and EZH2 to demonstrate the value of combination therapy with anti-CTLA-4 and CPI-1205, supporting the movement of the combination of EZH2 inhibition with anti-CTLA-4 into clinical trials.
by Brynn Vessey



