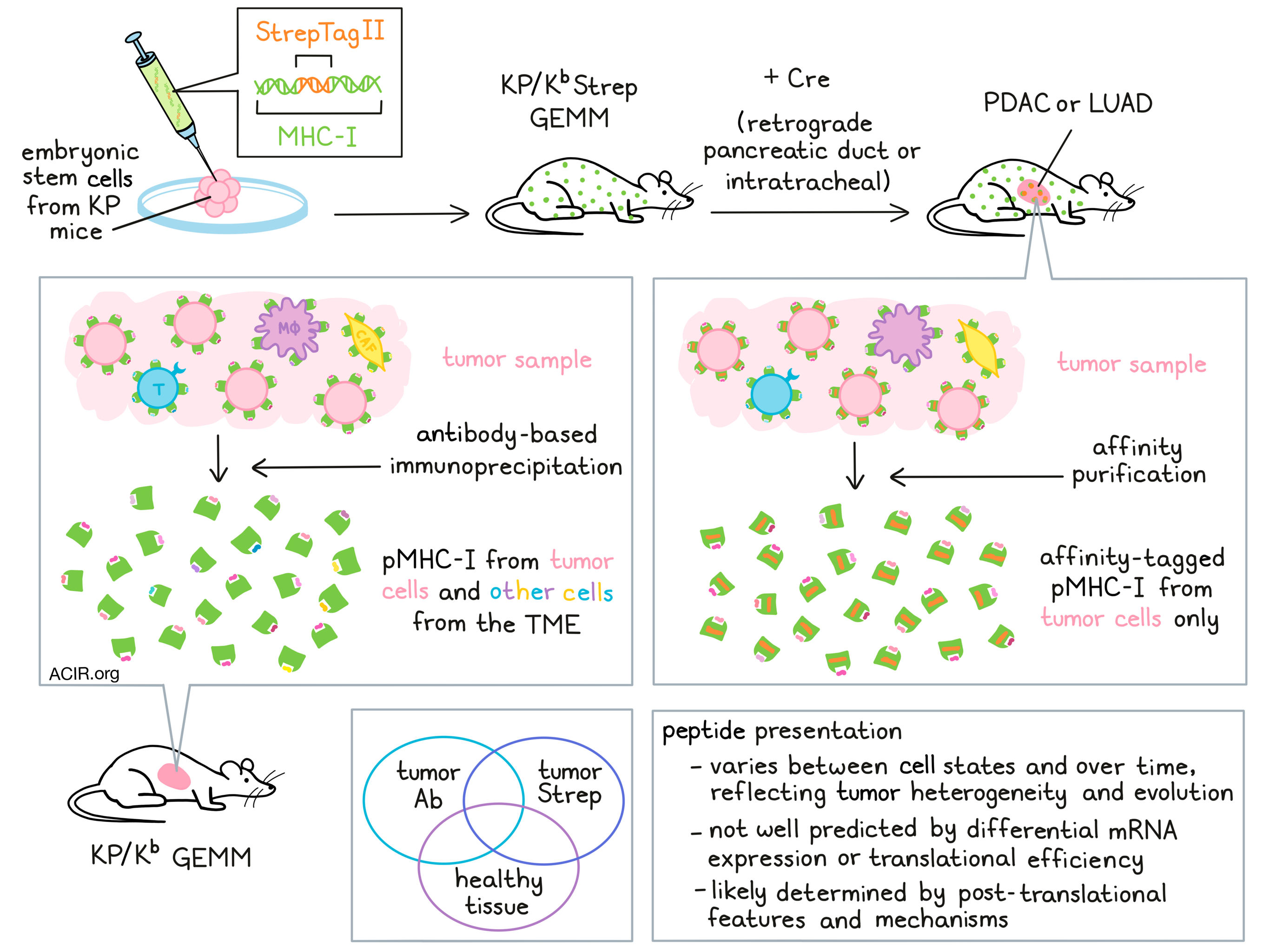
The immunopeptidome – the repertoire of peptide:MHC (pMHC) molecules presented on the surface of a cell – can reveal a lot about cancer cells and potential antigenic targets for cancer immunotherapy. However, current approaches to study the immunopeptidome in vivo are limited to heterogeneous bulk tumor or lysate samples, which do not allow for specific evaluation of the immunopeptidome of cancer cells in vivo. To overcome this hurdle, Jaeger et al. developed a novel genetically engineered mouse model (GEMM) that enables specific purification of pMHCs from cells of interest. Their findings were recently published in Nature.
To develop this mouse model, Jaeger et al. developed a Cre-inducible exon encoding a highly specific affinity tag, StrepTagII, to be inserted into the gene encoding MHC-I. This KbStrep allele was then targeted into embryonic stem cells derived from KP mice. In this new model, administration of Cre recombinase could be used to initiate activation of oncogenic KrasG12D, biallelic deletion of Trp53, and activation of StrepTagII. This strategy effectively tagged MHC-I molecules specifically on cancer cells to allow for affinity purification of pMHC-I from autochthonous tumors. This strategy did not interfere with typical PDAC development or presentation of peptides, and the model was validated both in vitro and in vivo.
Next, Jaeger et al. applied the KP/KbStrep model to generate autochthonous lung adenocarcinoma (LUAD). To evaluate the efficacy of this model in profiling the immunopeptidome, the team compared their novel system of cancer-cell-specific StrepTactin affinity purification from KP/KbStrep tumors against the use of traditional antibody-based immunoprecipitation to isolate H2-Kb peptides from healthy lungs or 16-week tumor-bearing lungs. Cancer cell-specific StrepTactin affinity purification was also performed on KP/Kb wild-type tumors as a negative control. StrepTactin affinity purification identified 438 peptides specific to tumors, 192 of which were not identified by antibody-based immunoprecipitation. While antibody-based immunoprecipitation also identified 104 peptides that were not identified by StrepTactin affinity purification, antibody purification is not cell-type specific, so there was no way of telling whether these unique peptides came from cancer cells.
To better understand how the immunopeptidome reflects specific cellular identities or cellular states, the researchers derived gene signatures from peptides identified in healthy lungs (normal), through antibody purification from tumor-bearing lung tissue (Ab), or through affinity purification of MHC-I in KP/KbStrep tumors (Strep), and fit those signatures to scRNAseq data from normal lung cells. The Strep signature showed strong enrichment for the alveolar type 2 (AT2) cell phenotype, consistent with the mechanism of tumor initiation. Further, the Strep signature showed stronger enrichment for the AT2 phenotype than the Ab signature did, suggesting that peptides isolated from bulk tumor lysates are contaminated with peptides from other cells in the tumor microenvironment (TME).
Given recent evidence suggesting that KP LUAD tumors progressively lose AT2 identity, Jaeger et al. created a mouse model in which StrepTagII was expressed in AT2 cells in healthy lung tissue. Using affinity purification, the researchers confirmed that the AT2 immunopeptidome strongly reflected the AT2 cell identity. Next evaluating the immunopeptidome of LUAD cells across different time points – 8 weeks (early), 12 weeks (mid), and 16 weeks (late) – the researchers found that the immunopeptidome progressively diverged from normal, losing the AT2 signal, and instead gaining resemblance to club and bronchioalveolar stem cells, suggesting that the immunopeptidome reflects the histological changes in tumor cells over time.
Exploring further, the researchers applied AT2, early, mid, and late peptide-derived gene signatures to published scRNAseq data from normal AT2 cells and KP tumors throughout progression. Here, the late signature was less correlated with AT2 cells and more reflective of the phenotypic plasticity previously observed in late-stage KP tumor evolution. The late peptide signature was also correlated with increased inflammatory cytokine signaling and decreased Myc signaling, metabolic processes, and epithelial-to-mesenchymal transition. In the metastatic cluster of KP tumor cells, H2-K1 expression was low, and was lowest in the late signature, suggesting that antigen presentation may vary across cell states and over time. This was further supported by multiplexed immunofluorescence imaging of late-stage tumors, which revealed intra- and inter-tumoral heterogeneity in MHC-I presentation.
To study whether changes in the TME alter the immunopeptidome, the researchers showed that depletion of CD8α+ immune cells from the TME decreased the number of unique MHC-I peptides. Similarly, inducing CD8+ T cell infiltration with agonist CD40 and FLT-3 ligand increased the number of previously unknown peptides, suggesting that antigen presentation is dynamically responsive to inflammatory cues in the TME.
Next, Jaeger and colleagues compared bulk RNAseq data against unique peptides from KP tumors, and found that genes encoding LUAD-unique peptides exhibited varied expression across timepoints, and that this did not correlate with either mean mRNA expression levels or predicted peptide affinity. Further, using transcripts that were upregulated at different stages of tumor progression to predict the affinity of possible peptides only identified 39 out of 312 total 8mers and 9mers on their LUAD-unique peptide list. Similarly, prediction of peptides based on protein synthesis (Ribo-seq) identified only 4 out of 312 empirically identified LUAD-unique peptides. Looking at post-translational features of proteins, the researchers found differences in source protein localization and a trend towards decreased protein half-life in source proteins that gave rise to LUAD-unique peptides. Inhibiting HSP90 to perturb post-translational processes resulted in an increase in the number of specific MHC-I peptides Identified, suggesting that the immunopeptidome can be reshaped through post-translational mechanisms.
Evaluating new tumor epitopes identified in the KP/KbStrep model, the researchers found 135 tumor-specific peptides that were recurrently expressed in tumors, but not in any healthy tissue in previously published data (tumor-specific antigens; TSAs). They also found another 147 peptides that were only found on one tissue (other than lung), which could represent possible tumor-associated antigens (TAAs). To study immunogenicity, the researchers then incorporated 3 tumor-specific antigens and 5 tumor-associated antigens into a peptide dendritic cell vaccine and delivered it into healthy mice. Three of the peptides were immunogenic (one TSA and two TAAs). Importantly, 2 out of these 3 peptides were not presented by cells in vitro, and none of the peptides would have been identified as priorities through differential analysis of RNA or ribosome protected fragments (RPF) compared to normal AT2 cells. Next, the researchers vaccinated mice bearing 5-week KP tumors. Here they were able to detect CD8+ T cells that recognized a peptide derived from one of the 3 immunogenic tumor antigens.
Overall, these results show that GEMM can be effectively used to study the in vivo immunopeptidome, which is complex and dynamic, and changes in response to tumor evolution and environmental stimuli. Additionally, these studies suggest that peptide presentation is not closely related to mRNA abundance or translational efficiency, and is instead may be driven by post-translational features and mechanisms.
By Lauren Hitchings

MEET THE RESEARCHER
This week, first author Alex Jaeger answered our questions.
What was the most surprising finding of this study for you?
One of the most surprising outcomes of this study was the identification of peptides that were differentially presented on cancer cells versus normal tissue with no apparent difference in expression at the RNA level. The process of antigen presentation is multilayered and complex, and the field has known for quite some time that the correlation between RNA, protein, and MHC-I peptides is not very strong. However, our results suggest that there may be numerous peptides that are presented in vivo that are unpredictable by transcriptomic analysis, and that many of these peptides may be immunogenic and therapeutically relevant.
What is the outlook?
Continued investigation into antigen presentation in the context of the tumor microenvironment will be critical for designing the next wave of peptide:MHC-specific therapeutics. In clinical samples, significant technological advancement is needed to ask these questions. However, using the novel mouse models described in our study, we can begin asking questions about how physiological perturbations influence the process of antigen presentation in vivo, including immunomodulatory treatment, metabolic perturbation, aging, microbiome alterations, and much more. Ultimately, these studies will improve our understanding of which peptides are presented in a given context, and perhaps identify strategies to dictate which peptides cancer cells will present. We hope to apply these mechanistic insights toward the identification of actionable antigens that can be used for therapeutic strategies in cancer patients, including cancer vaccines.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently started my independent lab at Moffitt Cancer Center in Tampa, Florida, and we also welcomed our second baby boy in May. Starting an independent lab and being a parent can be very challenging and it can sometimes feel like you lead two separate lives in the lab and at home. Although, sometimes worlds collide like when I get pictures of my 2.5 year old son wearing a lab coat in preschool “science class”. Through this process, I have learned that I don’t have to sacrifice being a good scientist to be a good parent. For me, setting the right mindset and priority structure is essential to balance both worlds.



