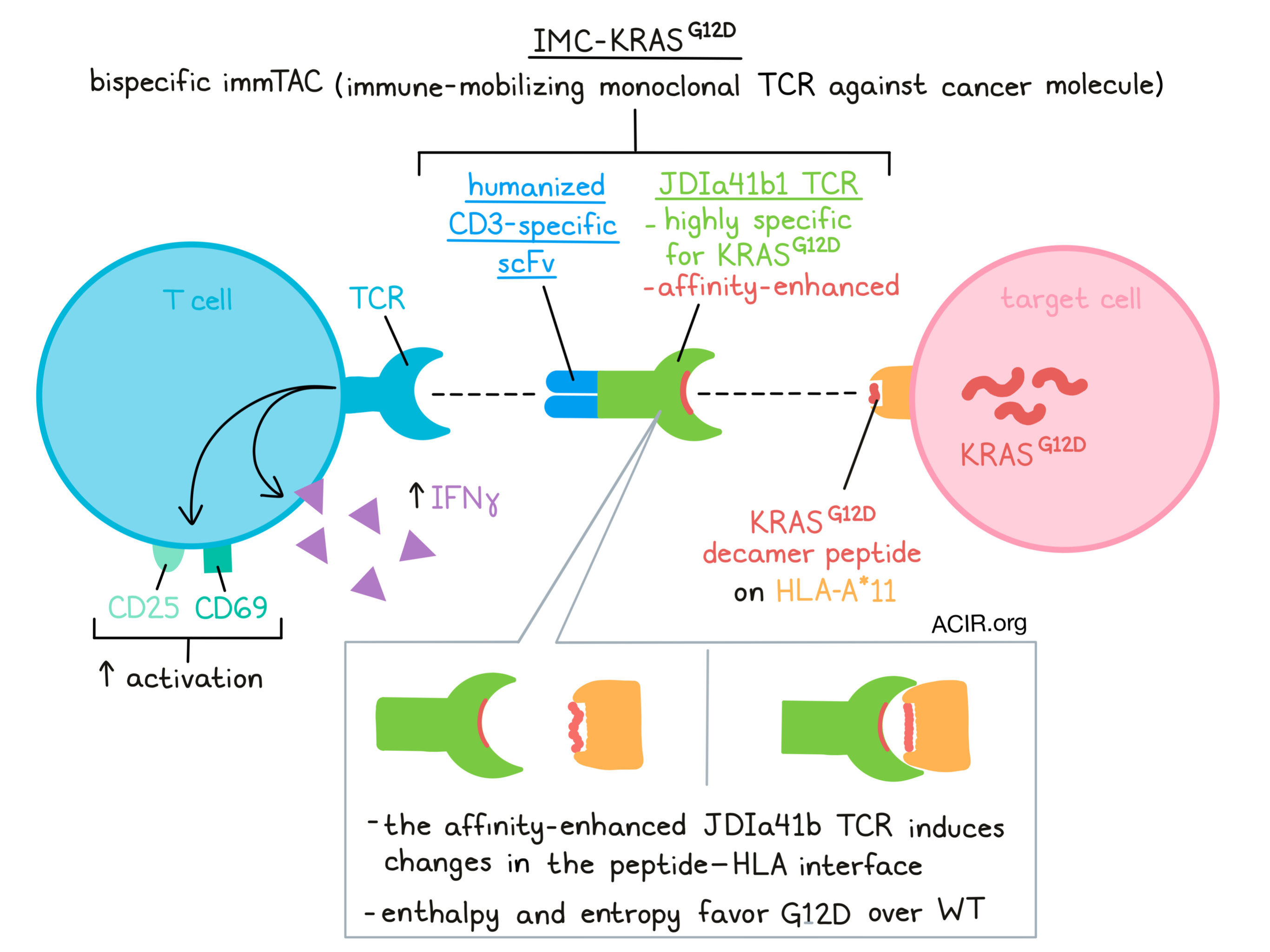
The oncogene KRAS is mutated in many cancer types, with the most common mutations being G12D, G12V, and G12C. As an intracellular protein, it is inaccessible to antibody-based therapy strategies. However, T cell receptors can detect the mutated proteins presented as peptide-HLA (pHLA), suggesting that these proteins can be targeted with TCR-based therapeutics. Poole et al. isolated a human TCR specific to the KRASG12D decamer peptide presented in the context of HLA-A*11 and analyzed its characteristics to further understand the selectivity of the TCR. Then, using this TCR as the targeting arm, they created a bispecific T cell-engaging ImmTAC (Immune mobilizing monoclonal TCR against cancer) molecule and assessed the functionality of this therapy in vitro, in work that was recently published in Nature Communications.
The KRASG12D-specific TCR (JDI TCR) was detected in human peripheral blood mononuclear cells (PBMCs) from a healthy HLA-A*11+ donor. T cells were transduced with the JDI TCR and cocultured with peptide-pulsed HLA-A*11+ acute lymphoblastic leukemia SUP-B15 B cells. When cells were pulsed with KRASG12D decamer peptide, but not KRASWT, T cells produced IFNγ. To assess selectivity, the binding of soluble JDI TCR to peptides from other RAS superfamily members with high similarity in amino acid sequences and to pools of self-peptides from ubiquitously expressed genes was assessed. No binding was detected to these other pHLA complexes, suggesting high selectivity.
To improve targeting of antigens that are presented at low levels, the TCR affinity can be strengthened. The researchers enhanced the affinity of the specific JDI TCR using NNK randomization of complementarity-determining regions (CDR), and used affinity variant phage libraries to select for TCRs with improvements in both affinity to G12D and in the G12D/WT affinity window.
To better understand the molecular basis of TCR selectivity, the researchers solved the crystal structures of the obtained affinity-enhanced JDIa41b1 TCR in complex with the HLA–peptides, which showed an almost identical conformation of the TCR and pHLA in the WT and mutant G12D complexes. However, there were differences between how the peptides interacted with the HLA, with the larger aspartate residue forming contacts with the HLA groove not possible with the smaller glycine, which may result in a more stable epitope when bound to the TCR.
To investigate the mechanism behind the affinity differences between mutant and WT complex for the JDIa41b1 TCR, the structures of both pHLAs without bound TCR were solved and compared to the conformation of the peptide in the JDIa41b1 TCR bound form of each complex. JDIa41b1 binding induced a shift in both peptides, switching the orientation of the central residues (4-6) from facing away from the HLA (in pHLA alone) to toward the HLA-F-pocket when the JDIa41b1 TCR was bound. This allowed the KRASG12D peptide to form the additional bonds with the HLA groove.
Solvation states and thermodynamics may play an essential role in defining the specificity and affinity of a TCR to antigens. Therefore, the researchers next assessed thermodynamics by performing surface plasmon resonance (SPR) over a range of temperatures to assess biomolecular binding. This revealed that JDIa41b1 TCR interactions with both pHLAs were enthalpically driven, with a greater favorable value for the interaction with KRASG12D, suggesting a larger gain in electrostatic interactions. Conversely, both TCR–pHLA complexes were entropically unfavorable, with the G12D structure being more unfavorable. To further assess the factors playing a role in the energetic differences, the researchers performed molecular dynamics (MD) simulations. A difference was found in the surface electrostatic potential of HLA-KRASG12D and -KRASWT around the mutation site and the TCR interaction zone.
Enhancing the TCR affinity may result in changes in the interface with pHLA, and the introduced mutations must maintain or enhance selectivity. To assess this selectivity, the researchers selected a third-generation affinity-enhanced JDI96b35 TCR and panned it against a high-complexity HLA-A*11 pHLA library using a single-chain trimer format displayed on phage. After three rounds of panning, 452 peptides were identified and used to create a peptide specificity profile, from which 20 self-peptides were identified that could act as structural mimetics of the KRASG12D peptide. This panel was then used to assess the binding of the JDIa96b35 TCR to peptide–HLA complexes other than KRASG12D. No binding was detected for 18/20 of the pHLAs, while two bound with weak affinity, suggesting high selectivity for KRASG12D.
Poole et al. then manufactured a bispecific ImmTAC molecule with the affinity-enhanced JDIa96b35 TCR fused to a humanized CD3-specific scFv (IMC-KRASG12D). ImmTAC molecules can directly bind their pHLA targets on tumor cells, redirecting and activating T cells to target tumor cells. Unstimulated PBMCs were incubated with SUP-B15 cells loaded with decamer KRASG12D or KRASWT in the presence of IMC-KRASG12D. In the cocultures containing KRASG12D, IFNγ was produced, and activated CD25+CD69+ T cells were detected.
Next, healthy donor immature dendritic cells (iDCs) electroporated with mRNA encoding WT or G12D KRAS were cocultured with autologous T cells and IMC-KRASG12D. The presence of IMC-KRASG12D resulted in increases in IFNγ production in cultures transfected with KRASG12D. Similar results were seen with peptide-pulsed untransfected iDCs. CRISPR/Cas9 was then used to introduce the KRASG12D mutation and/or HLA-A*11 into the pancreatic adenocarcinoma cell line PSN-1 to assess the specificity of IMC-KRASG12D. IFNγ was produced only in cocultures containing PSN-1 cells expressing both KRASG12D and HLA-A*11, indicating specificity for KRASG12D presented in the context of HLA-A*11. T cell assays were performed with cocultures containing IMC-KRASG12D and a series of target cell lines, including HLA-A*11+ cancer cell lines with various KRAS mutations and multiple healthy donor cell lines. Incubation of the CL40 colon carcinoma cell line containing the KRASG12D mutation resulted in the production of IFNγ, IL-2, and granzyme B, and directed T cell killing. Treatment with IMC-KRASG12D of the other cancer cell lines resulted in very limited to no IFNγ production, and no T cell killing was observed. IMC-KRASG12D also did not elicit IFNγ release when incubated with healthy cells.
These data suggest that an ImmTAC molecule targeting the oncogenic KRAS G12D mutation can function as a T cell engager that can specifically activate T cells and target cancer cells. This research shows proof of concept of targeting oncogenic neoantigens through the pHLA pathway.
Write-up by Maartje Wouters, image by Lauren Hitchings



