In a paper recently published in Cancer Immunology Research, Hartley et al. set out to explore how PD-L1 signaling affects the function of tumor-associated macrophages (TAMs), and in the process they discovered that PD-L1 blockade and PD-1 blockade confer distinct immunological effects.
To begin, the researchers examined bone marrow-derived mouse macrophages treated with an anti-PD-L1 antibody and found that the macrophages expressed high levels of PD-L1 and CD80, but not any detectable levels of PD-1 compared to an isotype control antibody. Culturing the macrophages with anti-PD-L1 increased their size, proliferation, and survival. The team also demonstrated in vitro that anti-PD-L1 induced activation of murine macrophages, as indicated by the upregulation of MHC II and the costimulatory molecule CD86. In addition, PD-L1 blockade led to a proinflammatory macrophage phenotype, as treated macrophages produced more TNFα and IL-12. Similar (though slightly less dramatic) effects were observed in human monocyte-derived macrophages.
To see if other PD-L1 ligands could reproduce the effect of the PD-L1 antibody, Hartley et al. treated the mouse macrophages with soluble PD-1 (sPD-1) or soluble CD80 (sCD80). As with anti-PD-L1, both treatments increased the size of the macrophages. Treatment with sPD-1 increased the expression of CD86 only, while sCD80 upregulated CD86, MHC II, and TNFα. Meanwhile, treatment with neither FcRII/III antibody nor CD11b antibody had any effect on the macrophages’ size, proliferation, activation, or survival; pre-treatment with the FcRII/III antibody did not abrogate the effect of anti-PD-L1 antibody treatment.
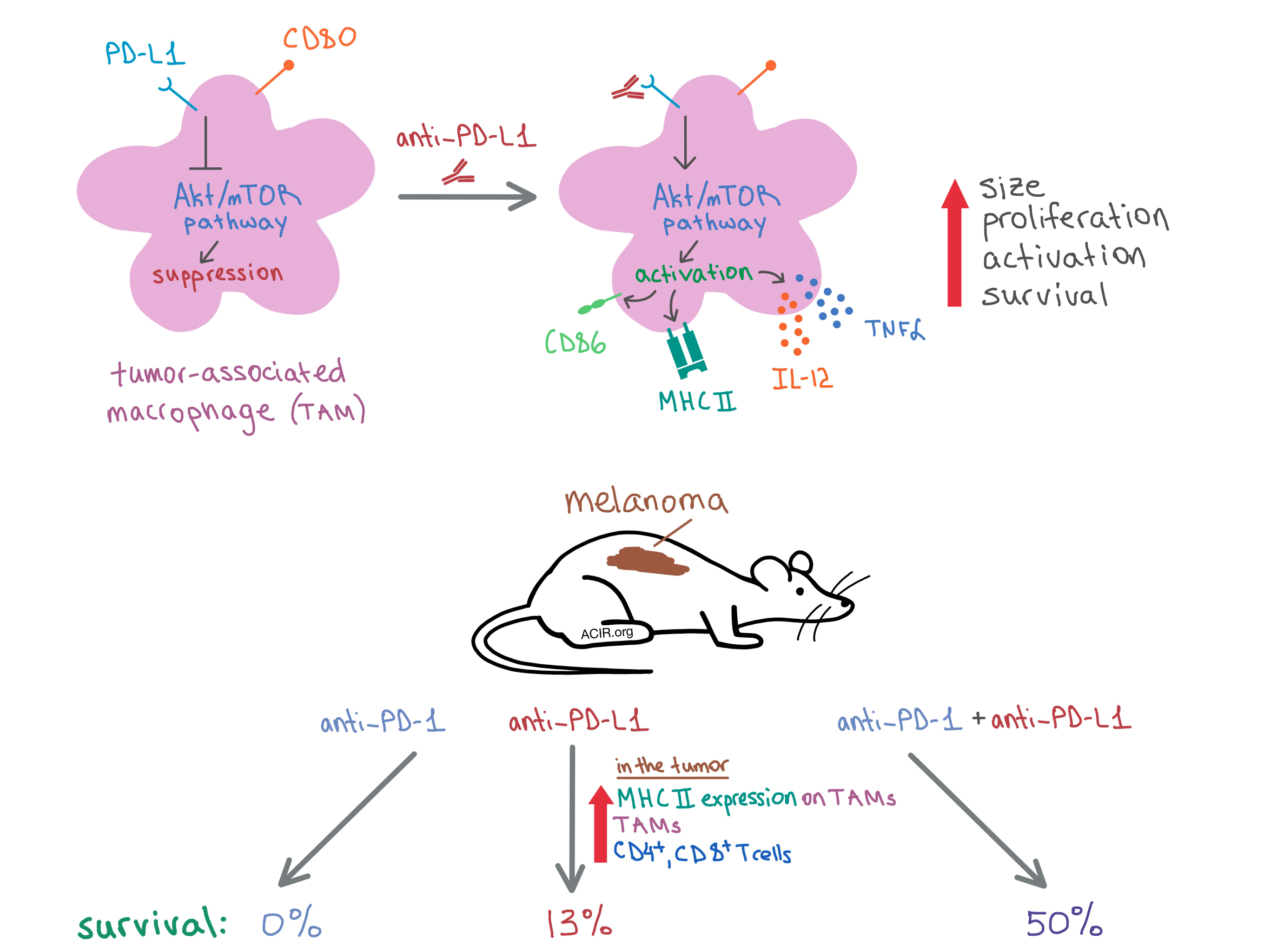
Digging deeper to discover the mechanism behind the anti-PD-L1 effect, the researchers examined macrophages from PD-L1-/- mice and found that these cells had increased proliferation and MHC II expression compared to macrophages from wild-type mice, indicating constitutive, negative PD-L1 signaling in wild-type macrophages. These results suggest that the anti-PD-L1 antibody interrupts PD-L1 signaling, leading to macrophage activation and other phenotypic changes.
Since PD-L1 signaling is involved in the mTOR pathway in mouse tumors, the researchers treated mouse macrophages with mTOR inhibitors rapamycin or torin2, which inhibited anti-PD-L1-induced proliferation, MHC II expression, and TNFα production. Meanwhile, anti-PD-L1 increased the levels of phosphorylated Akt and mTOR in macrophages. Together, these results indicate that constitutive PD-L1 signaling in macrophages inhibits the mTOR pathway, which regulates cellular metabolism, and that blocking PD-L1 removes the brakes from mTOR signaling, leading to macrophage activation.
The researchers then examined the anti-PD-L1-induced changes on a genetic level via RNAseq and transcriptome analysis. The data indicated that macrophages treated with anti-PD-L1 had upregulated inflammatory, survival, and proliferation pathways and downregulated anti-inflammatory and apoptosis genes, as well as pathways related to cellular processes.
Next, the team performed a series of experiments on mice with tumors to observe the effects of PD-L1 in vivo. In mice with B16 melanoma, anti-PD-L1 increased the infiltration of macrophages into the tumor, but not in spleen or lymph nodes. TAMs from treated mice had an increased expression of MHC II. More CD4+ and CD8+ T cells infiltrated the tumor post-treatment as well. In RAG-/- mice (lacking functional T cells) with PyMT breast carcinoma, the number of TAMs did not change with anti-PD-1 treatment, but CD86 and MHC II expression increased and tumor growth was delayed, indicating that anti-PD-L1 directly activates macrophages independent of T cells.
Since the results of the in vivo experiments demonstrated a T cell-independent antitumor response to PD-L1 blockade, Hartley et al. hypothesized that PD-L1 and PD-1 antibodies may have non-redundant antitumor effects. To that end, they treated mice with established B16 melanoma with either PD-L1, PD-1, or a combination of the two antibodies. They found that while monotherapies induced complete response in only 6% of mice, the combination treatment resulted in complete tumor rejection in 50% of animals. None of the anti-PD-1-treated mice survived to 68 days post treatment, while 13% of anti-PD-L1-treated mice and 50% of mice treated with the combination treatment were alive at that time.
Overall, Hartley et al. demonstrate that anti-PD-L1 treatment acts directly on TAMs to induce a proinflammatory phenotype and increase activation, proliferation, and survival via the activation of the mTOR signaling pathway. In addition, they show that PD-1 and PD-L1 antibody treatments result in distinct antitumor effects, and the combination of the two antibodies synergizes to produce a greater antitumor response than either therapy alone, suggesting another therapeutic opportunity.
by Anna Scherer



