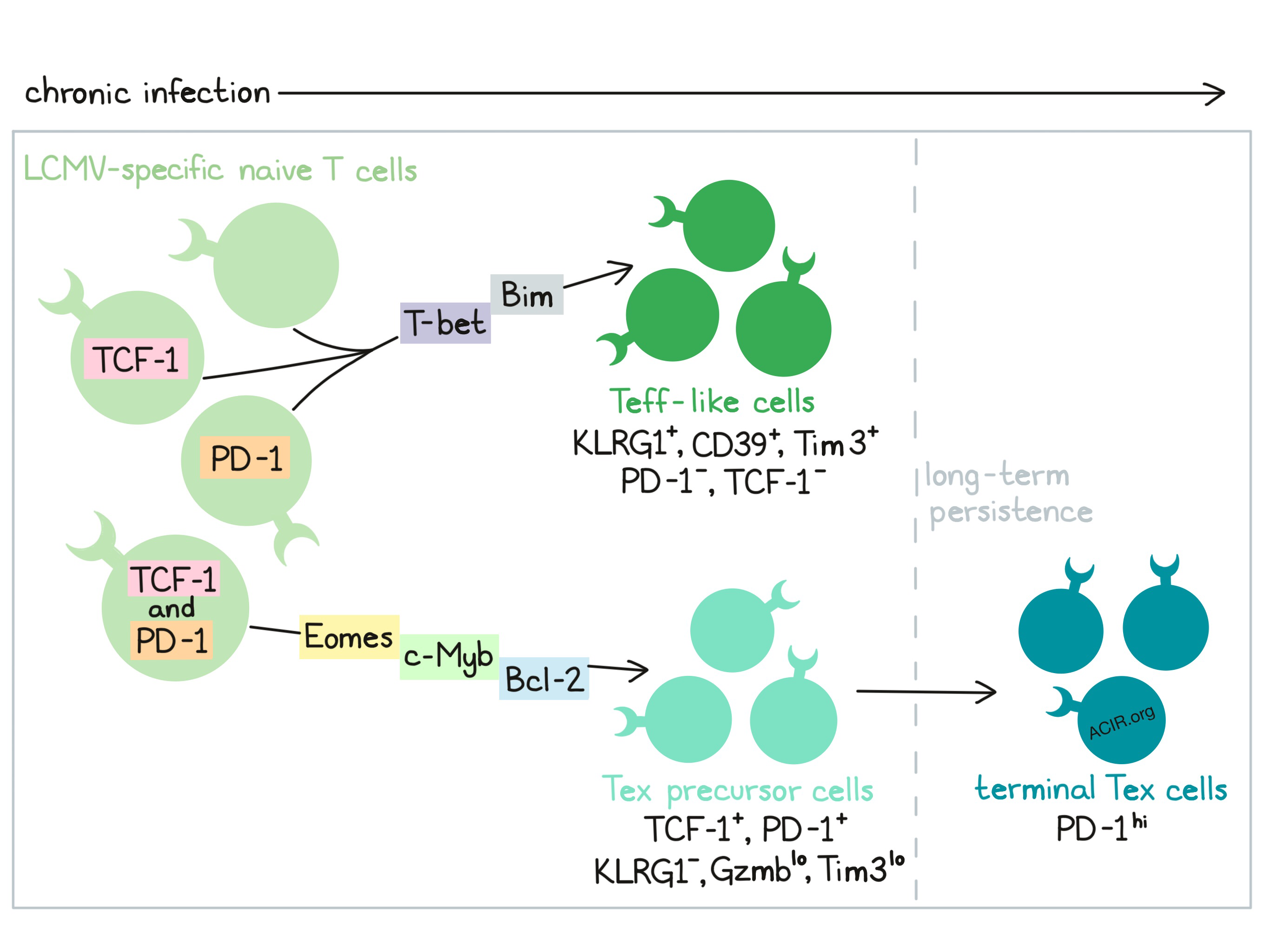
CD8+ T cell exhaustion plays a major role in chronic immune responses to both infection and cancer and is a major target of immunotherapy. To understand how and when exhaustion programs are initiated, Chen and Ji et al. analyzed gene expression data and transcription factor activity to define a binary fate decision early in chronic infection that separated effector from exhausted CD8+ T cell lineages. This decision point was largely mediated by the transcription factor TCF-1. Their results were recently published in Immunity.
To begin their studies, Chen and Ji et al. adoptively transferred LCMV-specific P14 cells into naive mice, infected the mice with acute or chronic LCMV, and performed single-cell RNAseq on viral antigen-specific CD8+ T cells at various time points. Visualization of the RNAseq data revealed five major clusters within responding cells 8 days after infection. Three of the clusters represented cells found almost exclusively in mice infected with the chronic strain of LCMV. One cluster in particular contained cells with high expression of Pdcd1 (encoding PD-1), Tcf7 (encoding TCF-1), Slamf6, and Bcl2, low expression of Gzmb (encoding granzyme B) and Havcr2 (encoding Tim3), and few markers of the cell cycle; these cells resembled TCF-1+ exhausted T (Tex) progenitor cells. Focusing on a set of exhaustion marker genes, the authors applied a lineage tracing approach to infer possible developmental pathways from the RNAseq data and found that naive T cells bifurcated into two branches early in chronic infection; one branch showed high expression of genes encoding PD-1 and TCF-1 and low expression of Gzmb and Klrg1, suggestive of a Tex progenitor-like branch. The other branch showed high expression of Klrg1 and genes encoding terminal differentiation markers Tim3 and CD39, suggestive of an effector T (Teff)-like branch.
Next, Chen and Ji et al. performed flow cytometric analysis based on expression of KLRG1 and PD-1, which were found to be nearly mutually exclusive. A large portion of KLRG1-PD-1+ cells expressed TCF-1, and those that did typically also expressed the transcription factor Eomes, showed low effector functions, were non-proliferative (based on Ki67), and persisted long-term, suggesting exhaustion. KLRG1-PD-1+ cells that lacked TCF-1 showed higher expression of the transcription factor T-bet. KLRG1+PD-1- cells showed increased CD39 and Tim3, and rarely expressed TCF-1, likely representing effector-like T cells. KLRG1+PD-1- cells were also found to decline slowly during acute infection, but rapidly during chronic infection, with minimal persistence. Investigating these patterns in persistence, the researchers looked at expression of anti-apoptotic Bcl-2 and pro-apoptotic Bim. They found that the KLRG1+ cell population had lower Bcl-2/Bim ratios, indicating high apoptosis in line with the observed low persistence. PD-1+ cells had higher Bcl-2/Bim ratios with PD-1+TCF-1+ showing the highest ratios, indicating low apoptosis, in line with longer persistence.
The researchers then adoptively transferred isolated cell subsets to experimentally define their properties and lineage relationships. They found that KLRG1-PD-1+Ly108+ cells (Ly108 being a surrogate marker for TCF-1) Tex precursor cells produced progeny at a high rate. This subset did not give rise to KLRG1+ cells, suggesting that early in chronic infection Tex precursor cells are distinct from KLRG1+ effector T-like cells and that these cell subsets are already on separate trajectories early during chronic infection.
Based on prior evidence that TCF-1 is involved in T cell exhaustion, the researchers explored its role in determining T cell fate. When Tcf7 was conditionally knocked out, the T cells mounted an initial response, but failed to seed a durable Tex cell pool, favoring branching towards the KLRG-1+ Teff-like fate rather than the PD-1+ Tex-like fate. Enforced expression of either of two TCF-1 isoforms repressed the formation of KLRG1+CD39+ or Tim3+CD39+ cells, increased the proportion of CD39-Ly108+ cells, promoted PD-1 expression, and fostered durability of the Tex cell population, suggesting that TCF-1 plays a role in the induction of exhaustion and the persistence of a Tex population. One of the TCF-1 isoforms was also found to repress KLRG1 and CD39 expression, suggesting that TCF-1 may also play a role in repressing the terminal effector phenotype early during chronic infection.
Investigating the relationship between TCF-1 and PD-1, Chen and Ji et al. found that while TCF-1+ cells late in chronic infection had lower PD-1 expression than TCF-1- cells, early in chronic infection TCF-1+ cells had high PD-1 expression. Knocking out PD-1 early in chronic infection increased the KLRG1+CD39+ and Tim-3+CD39+ cell populations while reducing TCF-1+ cells, suggesting that PD-1 plays a role in stabilizing the TCF-1+ Tex precursor population and in repressing the formation of terminal Teff-like cells.
To identify transcription factors, individually or as clusters, that were either correlated or anti-correlated with Tcf7, the researchers developed a computational approach called state transition interference predictor (STIP). STIP revealed that TCF-1 repressed transcription factor genes involved in Teff cell differentiation (Id2, Prdm1, Runx1) and promoted expression of transcription factor genes implicated in fostering exhaustion (Eomes, Batf, Nfatc1), suggesting that TCF-1 coordinated additional transcriptional circuitry.
Hypothesizing that TCF-1 might regulate T-bet and/or Eomes activity in developing Tex precursor cells, Chen and Ji et al. found that early in chronic infection, TCF-1+ Tex precursor cells expressed lower T-bet than TCF-1- cells. T-bet fostered the development of KLRG1+CD39+ Teff-like cells, promoted Tim3, and suppressed PD-1, suggesting T-bet promotes Teff differentiation, and that TCF-1 represses T-bet. Additionally, knockout of TCF-1 reduced expression of Eomes and c-Myb, accompanied by reduced Bcl-2. Eomes was found to play a role in the establishment and durability of Tex cells and c-Myb was found to antagonize Teff-like branch of differentiation and foster the Tex precursor cell population.
Overall Chen and Ji et al. defined a binary fate decision during chronic infection that guides effector (Teff) versus exhausted (Tex) CD8+ T cell differentiation. This differentiation is governed largely by TCF-1, which suppresses T-bet and the development of terminal KLRG1+ effectors, while fostering Tex precursors by promoting Eomes, c-Myb, and Bcl-2, which further support exhaustion and persistence. Formation of the TCF-1+ Tex precursor population is dependent on expression of PD-1. Understanding the early development of exhaustion in chronic infection serves as a useful underpinning to understanding checkpoint blockades and other possible immunotherapeutic strategies to support strong responses against chronic infections and cancer.
by Lauren Hitchings



