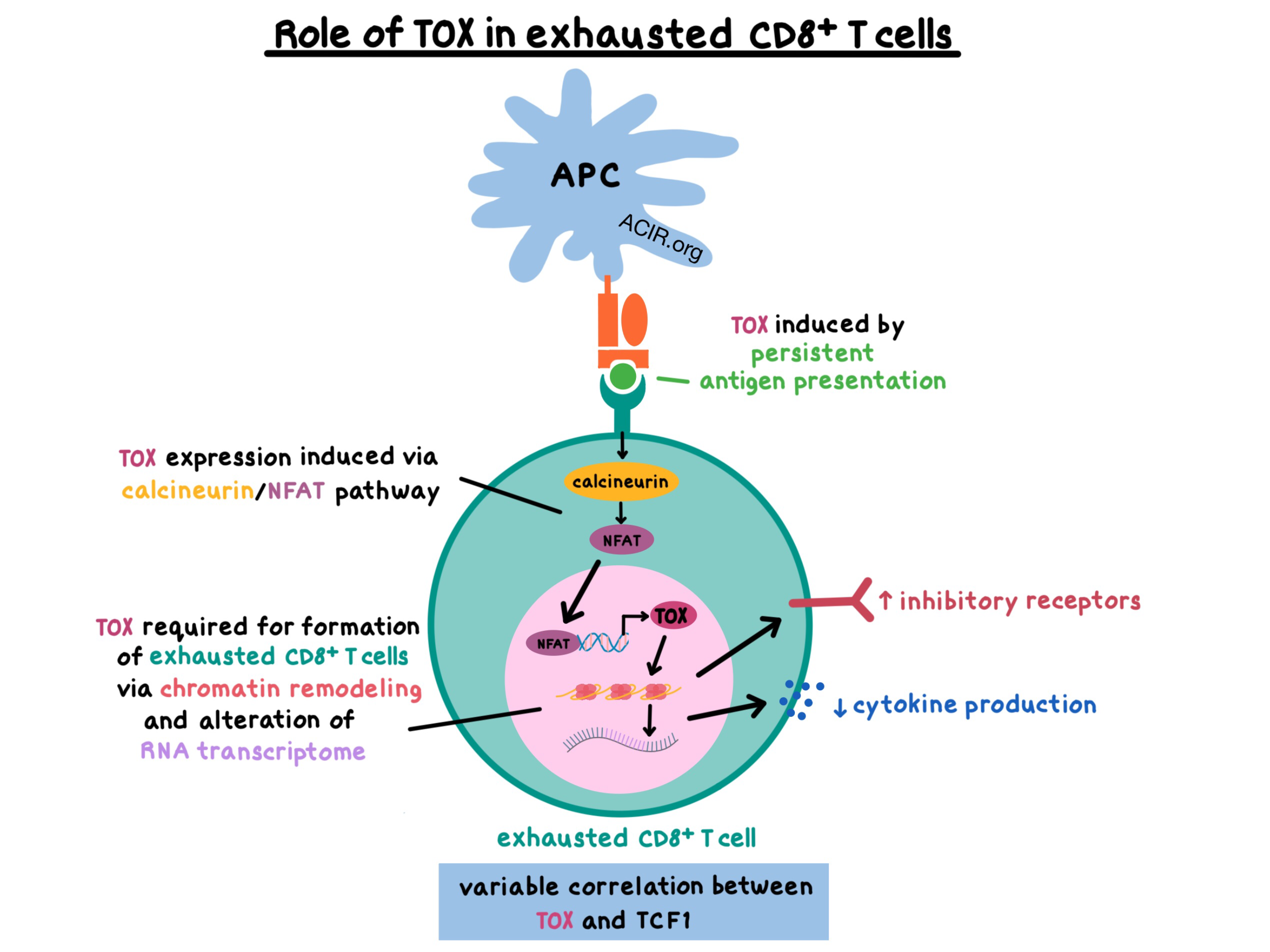
In this special feature, we summarize five recently published papers that explore the role of the transcription factor TOX in exhausted CD8+ T cells, which are common in cancer and chronic infections, and are the major target of checkpoint blockade. Although the research groups utilized different models (acute infection, chronic infection, tumors) and a variety of methods, their findings converged to indicate that:
- TOX preferentially plays a role in the setting of persistent antigen presentation, but not in acute infections
- TOX was required for the formation of exhausted CD8+ T cells via chromatin remodeling and alteration of the RNA transcriptome
- TOX induced high expression of inhibitory receptors and decreased the production of inflammatory cytokines
- TOX expression was induced by the calcineurin/NFAT pathway
A positive correlation between expression of TOX and the transcription factor TCF1 (a marker of progenitor-like CD8+ T cells) was mostly, but not universally, observed. The findings of each research group are presented in more detail below.
TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion (Khan et al., Nature)
Khan et al. analyzed the transcription data of virus-specific CD8+ T cells responding to an acute or a chronic LCMV infection and found that exhausted T cells were a distinct population from effector or memory T cells and that the Tox gene was the most differentially expressed gene associated with exhausted T cells. The TOX transcription factor was expressed only transiently and at low levels during an acute infection, but was persistently expressed at high levels in a chronic infection. Interestingly, expression of TOX diverged in vivo before viral load differences were observed in acutely versus chronically LCMV-infected mice. TOX was required for the formation of exhausted T cells and suppressed KLRG1+ effector T cell differentiation via chromatin remodeling. TOX was a critical regulator of exhaustion features, including the high expression of inhibitory receptors (such as PD-1, TIGIT, and LAG3) and transcription factors (Eomes, TCF1), impaired function, and decreased production of inflammatory cytokines. Mechanistically, TOX was initially induced by calcineurin via NFAT2, but its expression was ultimately sustained in exhausted T cells during chronic infection independently of calcineurin.
TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion (Seo et al., PNAS)
Seo et al. inoculated mice with human CD19-expressing B16-OVA melanoma and treated them with adoptively transferred CD8+ CAR T cells targeting human CD19. Tumor-infiltrating CAR T cells increased the expression of TOX and PD-1 over time. CAR TILs with a double deficiency of TOX and TOX2 transcription factors mediated tumor regression and improved survival in mice more effectively than WT CAR T cells or CAR T cells singly deficient in either TOX or TOX2. Seo et al. made similar observations to Khan et al. in terms of the effect of TOX on cytokine production, transcription factors expression (Eomes and TCF1), inhibitory receptors expression, and cytolytic activity of CD8+ T cells, and also demonstrated that TOX suppressed the expression of effector function genes.
Mechanistically, the expression of TOX, TOX2, and the nuclear receptor NR4A family members (transcription factors previously shown to be essential for induction of dysfunction), was induced by calcineurin via NFAT in the exhausted CD8+PD-1highTIM3high CAR TILs. NR4A and TOX positively regulated each other, and TOX, NR4A, and NFAT transcription factor families all contributed to the expression of inhibitory receptors (PD-1, TIM3, LAG3). Thus, Seo et al. showed that TOX and NR4A transcription factors were essential for the exhaustion transcriptional program in CD8+ T cells.
TOX is a critical regulator of tumour-specific T cell differentiation (Scott et al., Nature)
Scott et al. analyzed the role of TOX in autochthonous, SV40-T antigen-driven liver cancer and murine B16F10 melanoma models and in acute and chronic LCMV infection models using RNAseq and ATACseq and showed that Tox was highly expressed in dysfunctional antigen-specific CD8+ T cells but not in naive, effector, or memory T cells. Similar to the results published by Khan et al., TOX expression (regulated by NFAT) was low and transient in an acute infection, but persisted at high levels in a chronic infection and in tumor-specific CD8+ T cells during tumor progression. Interestingly, while high TOX expression correlated with high levels of inhibitory receptors and impaired cytokine production, it also correlated with low expression of TCF1 – an observation that differed from those of Khan et al. and Seo et al. TOX-knockout cells expressed high levels of the TCF1 gene, but were also enriched in apoptosis genes and failed to persist in the tumors. In vitro, enforced expression of TOX led to high inhibitory receptor expression but the T cells remained highly functional. TOX knockout of transferred cells in vivo did not increase inhibitory receptor expression but the cells were still dysfunctional. Together, these observations suggest uncoupling of function from modulation of inhibitory receptors. Tumor-specific T cells upregulated TOX and became dysfunctional, while non-tumor-specific bystander cytotoxic T cells remained TOXlow and functional, indicating that antigen stimulation leads to the induction of TOX and exhaustion.
Based on these results, Scott et al. suggest that TOX-induced regulation of exhaustion programs serves as a negative feedback mechanism to prevent T cell overstimulation and activation-induced cell death in a persistent antigen stimulation setting, such as cancer or chronic infection.
TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection (Alfei et al., Nature)
In murine T cells in the setting of acute, chronic, or chronic with low antigen load LCMV infection and in human T cells in human chronic hepatitis C virus infection, Alfei et al. found that TOX was highly differentially expressed in acute versus chronic infections, that induction of TOX depended on antigen quantity but not affinity, and that TOX expression correlated with persistent epigenetic programs of exhaustion. In chronic infection, T cells with a conditional deletion of the DNA-binding domain in the Tox gene (mutant-TOX) had decreased PD-1 expression, increased cytokine production, and elevated KLRG1 expression, indicating a more polyfunctional, effector-like phenotype and demonstrating that TOX was necessary for the generation of the dysfunctional phenotype. In vivo, TOX served to mitigate organ damage associated with the immune response to an infection.
Although the mutant-TOX T cells initially mediated enhanced viral control, this effect was transient, as the mutated T cells initially expanded but then quickly declined in quantity. Thus, TOX was required for T cell maintenance in the settings promoting T cell dysfunction, but not under conditions promoting effector phenotype. These observations prompted the researchers to analyze the expression of TCF1, a transcription factor required for T cell maintenance in chronic infections. Although the levels of TCF1+ T cell population were similar between WT and mutant-TOX T cells 8 days post infection, the mutant-TOX TCF1+ T cell population sharply declined by 20 days post infection. This is consistent with the observations made by Scott et al. who demonstrated that TCF1+ T cells with TOX knockout fail to persist in the long term under chronic antigen stimulation. TCF1+ T cells coexpressed TOX and PD-1 (but not TIM3). The researchers concluded that TOX promoted long-term T cell maintenance via induction of a dysfunctional phenotype and maintenance of the TCF1+ progenitor T cells (which also give rise to TCF1- T cells).
Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection (Yao et al., Nature Immunology)
Yao et al. utilized single-cell RNA sequencing (scRNAseq) to analyze virus-specific CD8+ T cells in chronic LCMV infection and identified a cluster of progenitor-like cells with Tox and Tcf7 (encoding TCF1) expression. Using scRNAseq and bulk RNAseq at different time points post infection, the researchers showed that the transcriptional programs of CD8+ T cells responding to acute versus chronic LCMV infection diverged before the peak of T cell response. By day 7 (near peak response time), progenitor-like “exhausted” and memory precursor cells had already diverged, although both subsets had similar expression of Tcf7. Analysis of T cell subsets revealed that Tox was coexpressed with Tcf7 in the progenitor-like cells but not in the memory precursor subpopulation. The terminally exhausted T cells also expressed Tox but at lower levels.
Knockout of Tox altered the differentiation of terminally exhausted T cells toward KLRG1+ short-lived effectors. On the other hand, TOX overexpression increased the persistence of virus-specific CD8+ T cells but did not change the frequency of the TCF1hi cells, indicating no skewing of differentiation toward a progenitor-like state. TOX also allowed terminally exhausted TCF1loCD8+ T cells to persist during a chronic viral infection. TOX overexpression led to increased expression of inhibitory receptors and decreased cytokine production, but did not affect in vitro cytotoxicity. Overall, Yao et al. demonstrated in vivo that cell-intrinsic TOX activity is required for the differentiation of progenitor-like TCF1hiCD8+ T cells and for long-term CD8+ T cell immunity in chronic viral infection.
Conclusion
All five research groups clearly demonstrated that TOX plays a crucial role in the generation of exhausted CD8+ T cells and in the maintenance of the exhausted phenotype (upregulated inhibitory receptors, decreased cytokine production, epigenetic modulation) in the context of persistent antigen presentation, whether in a chronic infection or in a tumor setting. The researchers from various groups also demonstrated that TOX is highly expressed in human cancers, including melanoma, non-small cell lung cancer, hepatocellular carcinoma, breast cancer, and ovarian cancer. Together, these results and observations suggest that modulation of TOX is a potential therapeutic target for the reversal of T cell exhaustion in human cancers.
by Anna Scherer



