Although PD-1/PD-L1 blockade has classically targeted T cells, some tumors with low MHC-I expression respond to this treatment, which suggests that other cell types are also inhibited by PD-1 and respond to PD-1 blockade. As PD-1 is expressed on natural killer (NK) cells in several cancers, and loss of MHC expression increases the tumor susceptibility to NK cell-mediated killing, Hsu and Hodgins et al. explored whether NK cells play a role in the antitumor effect of PD-1/PD-L1 blockade. The results were published in The Journal of Clinical Investigation.
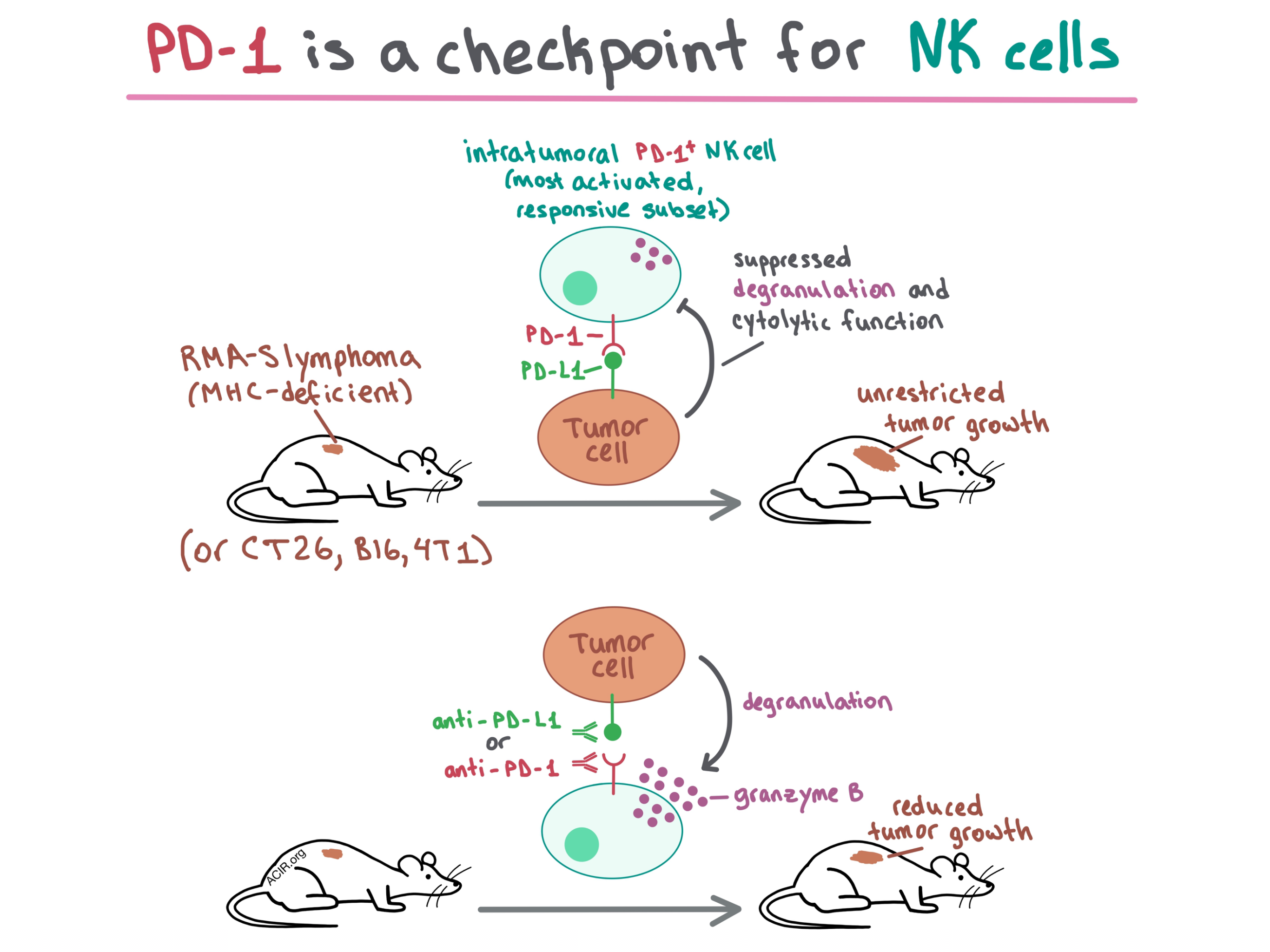
To begin, the researchers injected mice with RMA-S lymphoma cells, which have low MHC-I expression and therefore do not trigger a T cell response. Depletion experiments demonstrated that NK cells, but not T cells, played a crucial role in tumor control in these mice. When C57BL/6J mice injected with RMA-S-Pdl1 cells (RMA-S cells transduced with PD-L1) were systemically treated with anti-PD-1, growth of both smaller and more established tumors was decreased. This effect was abrogated when NK cells were depleted, demonstrating that anti-PD-1 triggered an effective NK cell response. Intratumoral injection of anti-PD-1 or anti-PD-L1 also reduced tumor growth. Together, these experiments showed that in MHC-deficient tumors, the efficacy of PD-1/PD-L1 blockade is dependent on NK cells.
Seeking to understand how PD-1 blockade affects NK cells, the researchers analyzed PD-1 expression and found that PD-1 was strongly upregulated on a large portion of intratumoral NK cells in RMA-S and multiple other tumor models. In vitro, PD-L1 expressed on tumor cells suppressed degranulation and cytotoxic function of PD-1+ NK cells. In vivo experiments in several tumor models confirmed that NK cells are inhibited by PD-1/PD-L1 ligation and can be rescued by PD-1/PD-L1 blockade.
The team then investigated whether NK cells would still play a relevant role when T cell response is added back into the equation. Depletion experiments in CT26 colon carcinoma tumors, in which PD-1 is upregulated on both NK and T cells, showed that PD-1/PD-L1 interactions inhibited both NK and CD8+ T cells in a comparable manner, that these two cell types were the major players in tumor rejection, and that they acted independently, at least in part. Anti-PD-L1 treatment slowed down tumor growth, but this antitumor effect was reduced when NK cells (or CD8+ T cells) were depleted. A separate experiment showed that NK cells preferentially killed PD-L1- tumor cells. Mechanistically, anti-PD-L1 treatment of mice with CT26 tumors increased the percentage of granzyme B+ PD-1+ NK cells, indicating restoration of function. Overall, the importance of NK cells in antitumor response, and their inhibition by PD-1, was demonstrated in 4 different tumor models: RMA-S lymphoma, B16-BL6 melanoma, CT26 colon carcinoma, and 4T1 mammary carcinoma.
Because PD-1 expression was heterogeneous on intratumoral NK cells in the tested tumor models, the researchers analyzed the different NK cell subsets. NK cells that were known to be more functionally responsive (e.g., they expressed self-MHC receptors [Ly49C+, Ly49I+, NKG2A+] or were in the R2 [CD11b+CD27+] maturation stage) were more likely to express PD-1. NK cells that expressed activation markers (Sca-1, CD69) were also more likely to express PD-1, suggesting that NK cell activation is related to PD-1 expression. Interestingly, PD-1 expression did not correlate with the stimulation by tumor cells, which suggests that PD-1 upregulation on NK cells may be driven by signals other than target cell stimulation. Further, using an ex vivo NK responsiveness assay, the team confirmed that PD-1+ NK cells were functionally more active than PD-1- NK cells.
Overall, Hsu and Hodgins et al. demonstrate in multiple tumor models that PD-1 is upregulated on the most activated and functionally responsive intratumoral NK cells, which explains why PD-L1 suppresses NK cell response despite the heterogeneous PD-1 expression on this cell type. The results of this study show that PD-1 is a checkpoint for NK cells, and that the efficacy of PD-1 blockade depends in part on inducing an NK cell-based antitumor response.
by Anna Scherer



