Last week, the ACIR team attended the AACR Tumor Immunology and Immunotherapy conference in Boston, MA. This week’s extensive special feature covers select sessions from the conference.
Opening Keynote and Special Lecture
Levi Garraway from Eli Lilly and Company opened the conference with an overview of the cancer-immunity cycle and how it relates to both response and resistance to immunotherapy. Despite the ever-increasing number of immune checkpoint blockade (ICB) therapies being approved by the FDA, the question of why the majority of cancer patients (particularly those with solid tumors) do not respond to immunotherapy remains unanswered. Additionally, as more and more ICB combinations are tested in clinical trials, the rationale for how to combine ICBs for optimal efficacy remains unclear.
Walking through the cycle, Garraway emphasized that both mutated neoantigens and non-mutated antigens are critical contributors to ICB response, and that they may be unleashed by other types of treatments, such as chemotherapy. Equally important is antigen presentation – the loss or induction of which could be mechanisms of ICB resistance or response, respectively. Another important factor is trafficking of T cells to the tumor, and Garraway suggested that studies combining PD-L1 blockade with an anti-angiogenesis drug are worth pursuing. Blockade of the PD-1/PD-L1 checkpoint signaling axis continues to be a staple in mono- and combination-immunotherapy trials, but it’s important to remember that PD-L1 expression is dynamic, and thus it is an imperfect marker of treatment response. A large meta-analysis comparing anti-PD-1 to anti-PD-L1 therapies revealed small, if any, differences. T cell exhaustion is another immunotherapy resistance mechanism, and new approaches to prevent or reverse it still need to be pursued. Finally, targeting the immunosuppressive tumor microenvironment would be a key goal of novel combinatorial immunotherapies.
In a special lecture that followed, Philip Greenberg from Fred Hutchinson Cancer Research Center talked about how to genetically engineer T cells with recombinant TCRs to eradicate cancer. One of the key factors for the success of this strategy is finding targetable antigens that are specific to the tumor and not expressed in normal tissue (to avoid toxicity). Another factor is ensuring that the antigen is correctly presented to the engineered T cells; this can be particularly problematic in long-term response, as tumors tend to adapt by progressing without expressing the targeted antigen following treatment. Thus, it is imperative to identify and target more than one tumor-specific antigen. To improve T cell activity, Greenberg discussed the design of ImmunoFusion Proteins, which contain an extracellular domain recognizing a target on tumor cells and an intracellular stimulatory tail so that recognition activates the T cell. Finally, new research is exploring concurrently administering engineered CD4+ T cells together with engineered CD8+ T cells, as the former play an important role in mediating the antitumor response of the latter.
In the last talk of the day, James Allison from the University of Texas MD Anderson Cancer Center discussed new developments and opportunities in ICB cancer immunotherapy. The blockade of CTLA-4 and PD-1 are two therapeutic approaches that have been studied extensively within cancer immunotherapy, and have gained multiple FDA approvals. Allison described in detail the mechanistic differences between the two approaches, pointing out that it is exactly these distinctions that may explain why the combination of the two therapies is so effective, noting that the mechanisms remain the same between highly immunogenic and poorly immunogenic tumors. Allison noted that it would be useful to identify and monitor specific subtypes of CD4+ and CD8+ T cells that contribute to the effect of both therapies in order to more accurately predict treatment outcome.
Synthetic Immunotherapy
Opening the plenary session on synthetic immunotherapy, Darrell Irvine from MIT explained his work on lymph node vaccination in combination with other immunotherapies. His lab has synthesized amphiphile-vaccines consisting of lipid polymer conjugates of peptides, proteins, or molecular adjuvants. The lipid polymer allows the vaccine to weakly bind to and essentially “hitchhike” on endogenous albumin. This hitchhiking strategy not only protects the peptide antigens from degradation, but increases delivery to the lymph nodes, significantly increasing the response to tumor antigens.
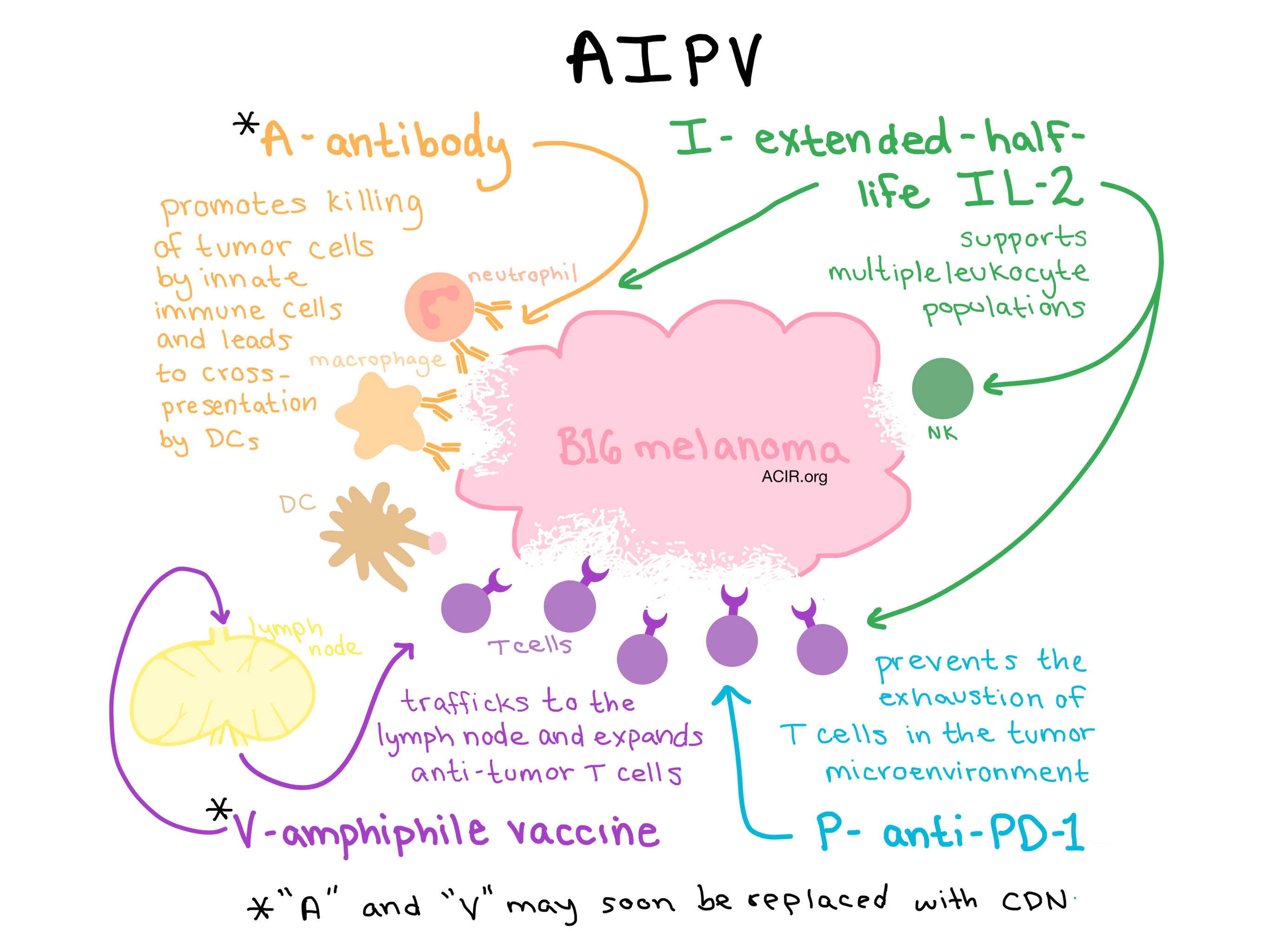
Combining the amphiphile-vaccine (“V”) with additional immunotherapeutic strategies - a tumor targeting antibody (“A”), an extended-life IL-2 (“I”) and anti-PD-1 (“P”) - together led to further enhancement of the immune response, including the eradication of large, established tumors in mouse models. This four-part “AIPV” therapy, while effective, is still rather complicated, so to simplify his strategy, Irvine is looking to replace the antibody and vaccine components with an in situ vaccine. He tried using the STING agonist CDN, which was effective, but induced a cytokine storm. He is now working on a carrier that would keep the CDN from spreading beyond the tumor microenvironment.
Following on with the theme of delivery, Matthias Stephan of Fred Hutchinson Cancer Research Center was next to speak. Stephan has published several papers this year that have caught our attention, including one on the use of biopolymers to co-deliver CAR T cells and STING agonists directly to a tumor site, another on the use of targeted nanoparticles to engineer CAR T cells in vivo, and a third on the many potential applications of using RNA nanocarriers to induce transient gene expression in target cells. In his talk, he explained results from the latter two papers, and reiterated his goal of minimizing the complexity and costs of targeted immune therapies by designing therapies that are simple to manufacture and can be stored long-term.
Yvonne Chen of the University of California Los Angeles considered how to take the pro-tumor factor TGFβ and transform it into an ally of CAR T cells instead. Because TGFβ is a soluble antigen, her group first looked at whether it was even possible for CAR T cells to recognize soluble antigens, and experiments with soluble CD19 proved that, in fact, they could. Interestingly though, CAR T cells were only responsive to dimerized/multimerized targets. Further experiments proved that it was not the dimerized ligands that were necessary, but dimerized CAR T cell receptors.
Moving forward with her original goal, Chen was then able to engineer CAR T cells with a surface domain that recognized TGFβ, and an intracellular signaling domain that stimulated an activation response. Manipulating the spacer lengths could tune the strength of the T cells’ responses to stimulation. Longer CARs were weaker activators, but ultimately yielded a longer-lasting responses as they were less affected by activation-induced cell death.
The final speaker of this session was Rohit Mathur of The University of Texas MD Anderson Cancer Center who spoke on the possibility of an off-the-shelf allogeneic CAR T cell therapy for multiple myeloma (MM) by targeting signaling lymphatic activation molecule F7 (SLAMF7 or CS1). Mathur proposed that the therapy could be generated for universal use by inactivating the TRAC gene (to reduce graft-versus-host disease) while knocking out SLAMF7 on T cells (to limit fratricide). He tested the efficacy of engineered UCARTCS1 cells against MM cell lines and samples and found that they specifically lysed target MM cells, and that both CD4+ and CD8+ UCARTCS1 proliferated in the presence of MM.
Neoantigens: The New Biomarkers
Opening the session on neoantigens, Jonathan Yewdell from the National Institute of Allergy and Infectious Diseases discussed how analyzing ribosomes using RiboSeq (a complicated but sensitive technique that precisely identifies RNA regions that are actively protected by the ribosome structure) can help identify peptides that appear to be over-represented in the peptidome, particularly peptides that arise from non-canonical protein translation. There is growing evidence that some MHC class I peptides arise from unexpected sources, including rapidly degraded defective ribosomal products (DRiPs), out-of-frame translation, CUG-initiated translation, and translation of introns and negative strands, and Yewdell suggests that such products are a rich source of antigenic peptides. Yewdell discussed the possibility that an “immunoribosome” exists that is responsible for unexpected peptide generation and plays a key role in immunosurveillance. Furthermore, tumors may modulate immunoribosomes to their advantage in order to escape detection by T cells.
In the following talk, Steven Rosenberg from the National Cancer Institute discussed how adoptive cell transfer (ACT) immunotherapy targets unique somatic mutations in cancer. (For more details on this concept, see our feature). Inspired by a study in which 55% of melanoma patients had objective response to ACT, Rosenberg and his team developed an approach based on deep exomic sequencing of tumors to identify immunogenic mutations, and then assess the reactivity of tumor-infiltrating lymphocytes (TILs) against the mutated neoantigens. The technique involved generating tandem minigenes that included a series of 25mers with the mutated amino acid in the middle flanked by 12 amino acids on both sides, and then transfecting the RNA into autologous antigen-presenting cells, which were then co-cultured with T cells.
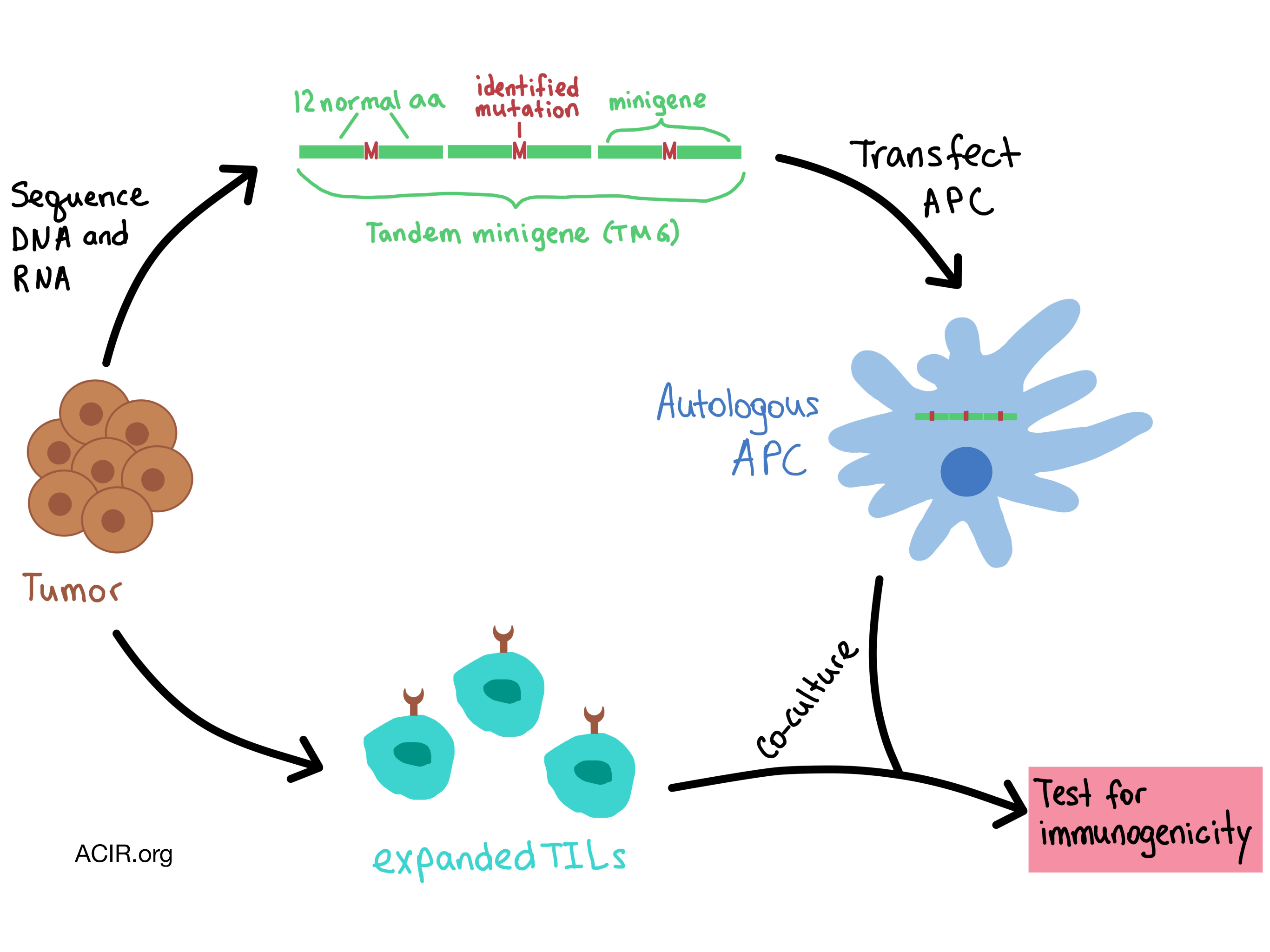
This approach was applied to the high-mutation-load melanoma tumors and to epithelial cancers with fewer mutations. In all cases, only approximately 1.5% of mutations turned out to be immunogenic, almost all were unique as a result of a random somatic mutation, and predicted affinities/rank mattered little in their occurrence. Rosenberg concluded that the recognition of random somatic mutations is the “final common pathway” that would explain response in most immunotherapies in solid cancers.
Catherine Wu from Dana-Farber Cancer Institute followed with a proof-of-concept clinical trial demonstrating that such mutations could be directly targeted with personalized neoantigen vaccines and have the power to increase patient response while minimizing toxicity. Wu and her team utilized next-generation sequencing approaches to identify immunogenic mutations and develop personalized vaccines for patients with stage III or resectable stage IV melanoma, and observed significant immunologic responses and intriguing clinical response data. Wu is now trying out this approach in patients with cancers that have lower mutation rates, such as glioblastoma multiforme. For a detailed discussion of Wu’s work on personalized neoantigen vaccines, see our feature.
In the final talk of the session, Claire Lhuillier from Weill Cornell Medical College hypothesized that radiation therapy could convert a tumor into an in situ vaccine by releasing tumor antigens (possibly even in poorly immunogenic tumors) and demonstrated the immunogenicity of two expressed neoantigens in irradiated tumors.
The inflammatory Tumor Microenvironment
Miriam Merad chaired this session and opened with a talk on the diversity of the innate immune system. She explained how tissue-resident macrophages arise from embryonic tissue, differing from monocyte-derived macrophages, which arise from bone-marrow progenitors. She described how tissue-resident macrophages resist stress, repair DNA, and contribute to tumor resistance through Treg induction and the shaping of fibrotic responses, suggesting that specific depletion of embryonic-derived macrophages would improve some immunotherapies. Finally, Merad discussed using a combination of single-cell RNA-Seq, CYTOF, and multiplex IHC, she tracked various immune cell populations through the early stages of tumorigenesis.
Shannon Turley of Genentech discussed another challenge plaguing immunotherapy researchers: getting CD8+ T cells to infiltrate the tumor bed. We know that responders to immunotherapy tend to have high levels of CD8+ effector cells infiltrating tumor beds, which can be described as inflamed. Alternatively, tumors with fewer infiltrating T cells are associated with poorer outcomes. Turley’s team analyzed gene expression patterns in these “immune excluded” tumors and found TGFβ and it’s receptor to be significantly upregulated and they tracked the mechanism down to activation of cancer associated fibroblasts. Combining anti-TGFβ with anti-PD-1 increased T cell infiltration of the tumor bed all the way to the centers of tumors. She also observed increases in effector T cell gene signature and decreased immune cell response to TGFβ with this combination treatment.
The remaining talks in this session focused primarily on dissemination and metastasis. Julio Aguirre-Ghiso of the Icahn School of Medicine at Mount Sinai explained the mechanism behind the dormant state of metastases that develop long after the primary tumor has been cleared, as well as the role of macrophages in early dissemination of these dormant cells. Nathan Reticker-Flynn of Stanford University discussed how cancers often first metastasize to the lymph nodes, promoting systemic immune tolerance that supports metastatic cells from the primary tumor. Dalit Barkan from the University of Haifa described how a fibrotic-like microenvironment with extensive deposition of Type 1 collagen and fibronectin promote the outgrowth of dormant tumor cells. Barkan further suggested that factors secreted by pro-resolving macrophages (F4/80+CD11blo), can subdue inflammatory and fibrotic responses and prevent the reactivation of dormant cancers.
Metabolic Regulation of Immune Responses
Mihai Netea from Radboud Institute for Molecular Life Sciences in the Netherlands discussed how myeloid cells can undergo long-term metabolic reprogramming after an infection or within the TME. Although myeloid cells are not usually thought to have an immunological memory, there is evidence that the innate immune system can mount an increased response to a secondary infection that follows priming during an initial infection. Initial infection can lead to either training (which may also be enhanced by vaccination) or tolerance. Trained myeloid cells switch to glycolysis and provide immune protection against lethal sepsis, while untrained cells maintain oxidative phosphorylation. The epigenetic changes observed in trained innate immune cells (e.g. macrophages) include histone modifications that enable chromatin to remain open, DNA methylation, modulation of miRNA, and expression of long-noncoding RNA.
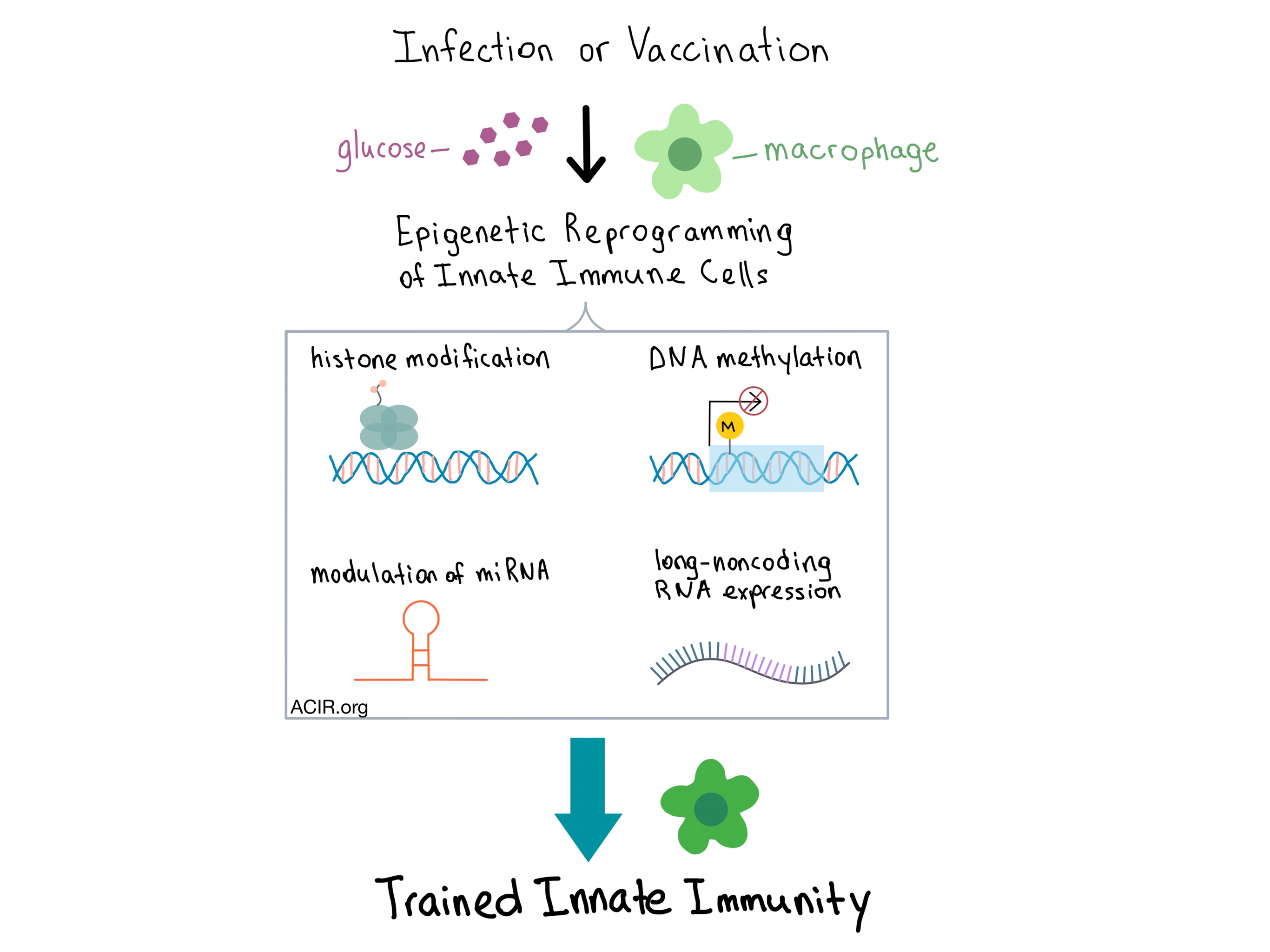
Within the TME, the tumor metabolites affect the function of macrophages by causing metabolic reprogramming towards a more inflammatory profile, which potentiates carcinogenesis. Thus, modulating the metabolism of the tumor and the immune cells could be a promising approach to explore in the treatment of cancer.
Other topics that were presented during this plenary session included: how the TME leads to a reprogramming of the Krebs cycle that results in altered cytokine production (by Luke O’Neill from Trinity College Dublin); using CRISPR-Cas9 to identify genes essential for cancer immunotherapy (by Nicholas Restifo from the National Cancer Institute - see our spotlight); and the metabolic adaptations that tumors undergo to confer resistance to immunotherapy in melanoma (by Ashvin Jaiswal from the University of Texas MD Anderson Cancer Center).
Checkpoints and Immunomodulation
Charles Drake from Columbia University Medical Center kicked off the session on checkpoints and immunomodulation by discussing the roles of Treg cells following radiation therapy in prostate cancer in mouse models. He noted that following radiation, Treg cells were the first to infiltrate the tumors, followed by myeloid cells. A combination of androgen deprivation therapy with anti-CTLA-4 therapy (using an antibody that is capable of Treg depletion) worked in synergy to cure mice. In a separate model, suppressive immune cells eventually built up following radiation therapy. Combining limited radiation therapy with anti-CTLA-4 led to durable long-term remissions in a large fraction of the mice, however, combining radiation with anti-PD-1 therapy was less successful.
One of the last talks of the conference was by Joshua Brody from the Icahn School of Medicine at Mount Sinai. While a high mutation load is typically associated with enhanced response to immunotherapy, Hodgkin’s lymphoma does not fit the pattern: it has relatively few mutations yet is still highly responsive to anti-PD-1 therapy. Brody and his team attributed this to the presence of dendritic cells (DCs) capable of cross-presenting tumor antigens. They hypothesized that checkpoint blockade can be limited by suboptimal cross-presentation by insufficiently activated DCs, and that if they could improve the recruitment, loading, and activation of cross-presenting DCs at tumor site, they could improve response to checkpoint blockade. Intratumoral FLT3 Ligand (FLT3L) led to significant recruitment of XCR1+, CLEC9A+, Langerin+ DCs and radiation enhanced the recruitment of CD103+ DCs.
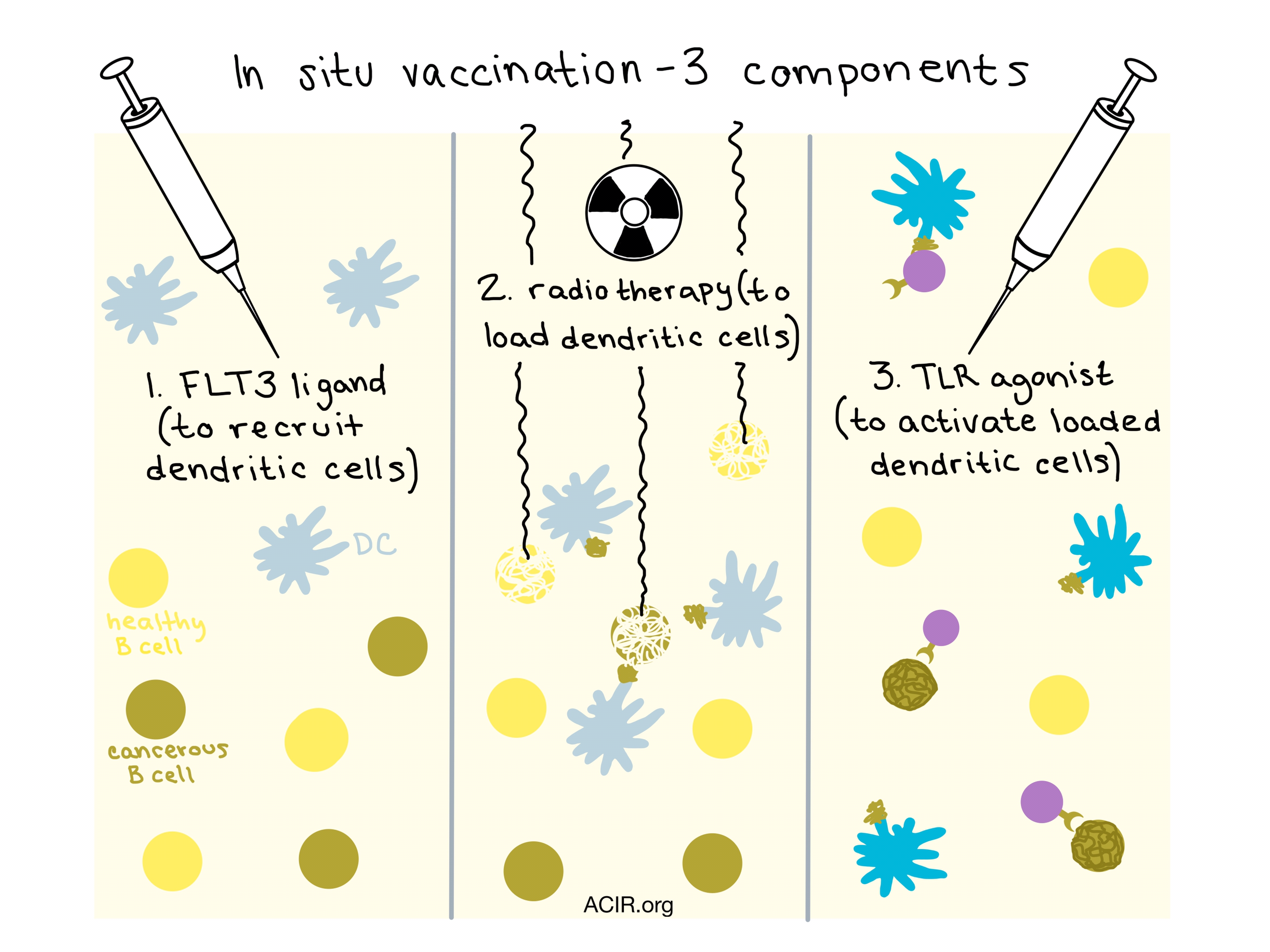
In an early clinical trial, Brody and colleagues tested an in situ vaccine with three components: FLT3L to recruit DCs, radiotherapy to load FLT3L-mobilized DCs with tumor antigens, and a toll-like receptor agonist (Poly-ICLC) to activate the loaded DCs for cross-presentation. They observed deep partial responses, including long-term regressions in distant, untreated tumors. Further, the vaccine was able to eliminate malignant B cells while sparing the healthy ones. Based on preclinical success in combining this vaccine with a PD-1 blockade, a new clinical trial of this combination has been set to open in 2018.
by Anna Scherer and Lauren Hitchings



