While immune checkpoint therapy (ICT) has successfully treated a wide array of cancers, the majority of patients do not benefit. This may, in part, be due to immune suppression within the tumor microenvironment (TME), relying on certain myeloid cell populations that can dampen T cell immunity and aid tumor progression. In work recently published in Cell, Molgora et al. identified the macrophage receptor TREM2 as a potential therapeutic target to modulate tumor-infiltrating macrophages and in turn bolster ICT efficacy.
TREM2, an Ig-superfamily surface receptor expressed on myeloid cells, mediates intracellular signaling and has previously been implicated in a variety of pathologies. TREM2+ macrophages have been observed in lung infection, atherosclerosis, and liver fibrosis, and TREM2+ microglia may be involved in Alzheimer’s disease. Additionally, TREM2+ macrophages have been detected in tumors. TREM2 can be upregulated upon exposure to cytokines such as GM-CSF or CSF-1, which are expressed in the TME, but its role in cancer progression remains unclear.
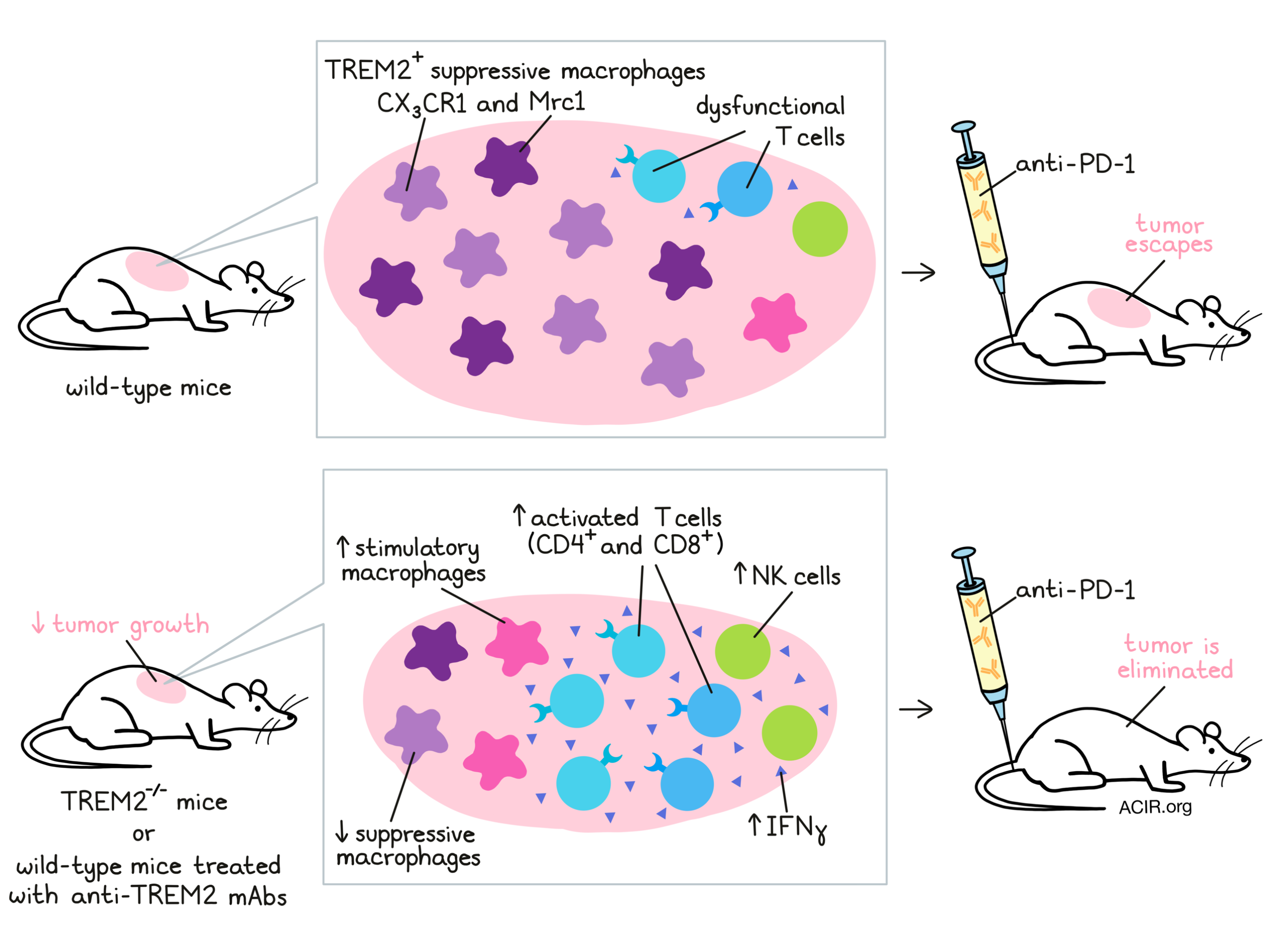
To investigate, Molgora et al. employed a germline TREM2-/- mouse model, characterizing the impact of TREM2 deletion on the growth of tumor lines known to have extensive myeloid infiltrates. In MCA/1956 carcinogen-induced sarcoma, MC38 colorectal cancer, and PyMT breast cancer, tumor growth was reduced in TREM2-/- compared to wild-type mice. Flow cytometry analysis 10 days after MCA/1956 tumor injection revealed alterations in intratumoral myeloid populations, increased T cell infiltration, and greater PD-1 expression on both CD4+ and CD8+ T cells in TREM2-/- hosts. These preliminary results indicated that TREM2 deletion both modified the TME myeloid compartment and supported T cell-mediated tumor control. Indeed, depletion of CD8+ T cells in TREM2-/- mice prevented this reduction in tumor growth.
For a closer look at the effects of TREM2 knockout on MCA/1956 immune infiltrates, the team next used single-cell RNA sequencing (scRNAseq). Of CD45+ cells sorted from tumors, 14 cell clusters were identified, corresponding to known immune cell types, including multiple subsets of macrophages, which all expressed TREM2. Similar to the results observed with flow cytometry, intratumoral myeloid clusters differed between TREM2-/- and wild-type hosts. Notably, CX3CR1 and Mrc1 macrophages – populations that expressed immunosuppressive genes – were decreased in TREM2-/- mouse tumors. Instead, a macrophage population expressing stimulatory genes (e.g. Cxcl9 and Cd83) predominated. The researchers also observed that the macrophage clusters with highest TREM2 expression were depleted in TREM2-/- mouse tumors, suggesting a role of TREM2 in maintenance or proliferation of these cells. In the lymphoid compartment, Molgora et al. found enrichment of T and NK cells in TREM2-/- mouse tumors, both of which increased expression of activation markers IFNγ and PD-1. Together, the scRNAseq results illustrated a more activating TME in the context of TREM2 deletion, involving stimulatory macrophages and enhanced T and NK cell activity. Supporting this hypothesis, these data were compared to a previously published dataset sequencing intratumoral macrophages after ICT (Gubin et al. 2018). Certain similarities were found, particularly a reduction in CX3CR1 and Mrc1 macrophages in both studies. However, ICT had induced stimulatory Nos2 and Rsad2 macrophages, a finding not mirrored in TREM2-/- mouse tumors. It appeared that TREM2 deletion and ICT could both remodel TME macrophage populations, but in distinct ways.
Following on from these observations, the authors next considered whether TREM2 deletion and ICT may serve as complementary therapies. As MCA/1956 tumors are responsive to anti-PD-1 therapy, a suboptimal ICT regime was found in which tumor growth remained relatively uncontrolled in wild-type mice. Strikingly, all TREM2-/- mice receiving the same anti-PD-1 regime rejected their tumors. These results were echoed in the MC38 tumor model. Using flow cytometry, the researchers found that suboptimal anti-PD-1 treatment in TREM2-/- mice improved total T cell and CD4+ T cell infiltrates versus in wild-type mice. Regardless of anti-PD-1 treatment, TREM2-/- mice again showed increased tumor infiltration by CD8+ T cells and increased PD-1 expression on infiltrating CD8+ T cells.
The next question was whether TREM2 could be targeted therapeutically, such as by a monoclonal antibody. To address this issue, the team developed an Fc-modified mAb against TREM2, which specifically bound TREM2+ cells, preventing ligand binding without depleting the cell target. In treating MCA/1956 tumors, anti-TREM2 alone was partially effective in controlling tumor growth, and the combination with suboptimal anti-PD-1 therapy eliminated all tumors. Interestingly, tumor growth was slowed more with anti-TREM2 treatment than by TREM2-/- hosts, which the authors suggest may potentially be due to compensatory mechanisms for macrophage survival emerging in the context of total TREM2 knockout.
An analysis of the TME following treatment revealed significant changes. The combination of anti-TREM2 and suboptimal anti-PD-1 altered the myeloid compartment, notably reducing CD11b+, Ly6C+MHCII-, and CD64+ cells at an early time point (day 10). By day 24, anti-TREM2 treatment alone led to decreases in Ly6CloMHCII- cells and CD206 expression (at this time point, no tumors were left to analyze in the combination group). T cells isolated from these tumors and stimulated with PMA/ionomycin ex vivo showed superior IFNγ and TNFα production over T cells from control antibody-treated tumors. Additionally, using scRNAseq, the researchers found that anti-TREM2, regardless of combination with suboptimal PD-1, induced Nos2+ macrophages and depleted Mrc1 macrophages. With both anti-TREM2 and anti-PD-1, CX3CR1+ cycling macrophages were also depleted. Broadly, these changes were consistent with the observations in TREM2-/- mice, as well as the previously reported remodeling of macrophages in the TME following ICT.
Finally, the team considered whether TREM2 could be targeted in human tumors. Through immunohistochemistry, TREM2 was seldom detected in patient normal tissue samples, but was expressed in a variety of cancers. Specifically, TREM2+ macrophages were identified in multiple human carcinomas, showed diverse morphologies, and co-expressed markers such as CD68, CD163 and CSF1R. TREM2+ macrophages localized exclusively within tumors, rather than peritumoral tissue where TREM2- macrophages were observed, and accumulated more within metastatic sites (lymph nodes, lungs, and liver) than in low-grade or benign lesions. Furthermore, an analysis of The Cancer Genome Atlas (TCGA) revealed correlations between TREM2 expression and worse overall survival in colorectal cancer, as well as poor overall and relapse-free survival in triple-negative breast cancer. In both cancer types, TREM2 expression correlated with myeloid gene signatures, but had independent prognostic capability for patient survival.
Overall, the researchers validated TREM2 as a therapeutic target capable of remodeling myeloid populations within tumors, supporting T cell immunity and boosting ICT for tumor control. Further, the identification of TREM2 as a potential prognostic marker in human cancers is encouraging to the clinical translation of this strategy, and suggests that anti-TREM2 antibodies could be added to the arsenal of therapies targeting suppressive myeloid cells in tumors. Future research into the mechanisms of how targeting TREM2 affects intratumoral macrophages, alone and in combination with established ICT, will continue to advance this therapy.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, first author Martina Molgora and lead author Marco Colonna answered our questions.

What prompted you to tackle this research question?
TREM2 is a macrophage receptor that was discovered in our lab years ago. We have extensively studied it in neurological disorders, because TREM2 is expressed in microglia, the brain-resident macrophages, and a mutant of TREM2 is associated with high risk for Alzheimer disease. Recently, we and others have noticed that TREM2 is also expressed in tumor-associated macrophages in both human and murine tumors. This is the reason why we pursued the potential role of TREM2 in regulating the immune response to cancer.
What was the most surprising finding of this study for you?
The most surprising finding is the activity of the anti-TREM2 antibody. Given that at least one anti-human TREM2 monoclonal antibody has been tested in a phase I clinical trial for Alzheimer’s disease, anti-TREM2 antibodies seem safe in humans and therefore they can be quickly tested in human tumors, alone or in combination with checkpoint blockade.
What was the coolest thing you’ve learned (about) recently outside of work?
Since we completed this work during the pandemic, we realized how important it is to be able to interact with our colleagues. Some of the most fulfilling aspects of our work involves team science. The lockdown emphasized how essential daily informal meetings are with the lab and the department, and we have been working to maintain those discussions as much as possible during these times.



