
In October, the ACIR team attended the virtual AACR Tumor Immunology and Immunotherapy conference 2021. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Tumor microenvironment and TME-targeted immunotherapy
Ping-Chih Ho
Greg M. Delgoffe
Saar Gill
Johanna A. Joyce
Jennifer L. Guerriero
Tumor immunity
Miriam Merad
Jérôme Galon
Nathan Reticker-Flynn
Judith Agudo
T cell therapies
Caroline Arber
Crystal L. Mackall
Systems biology
Grégoire Altan-Bonnet
Combination therapy
Jennifer A. Wargo
Jedd D. Wolchok
Tumor microenvironment and TME-targeted immunotherapy
Unconventional ER stress response pro-tumorigenic polarization and survival in TAMs- Ping Chih Ho - Ludwig Institute, Lausanne, Switzerland
Many of the tumor-associated macrophages (TAMs) in the tumor microenvironment (TME) are polarized to the immunosuppressive M2-like phenotype. Ping-Chih Ho and team investigated how the TME promotes the generation of these M2-like TAMs. They found that TAMs accumulate lipids and display ER stress response signatures (increases in spliced XPB1, IRE1 and BiP). ER stress has previously been reported in intratumoral dendritic cells, but it remains unclear what factors drive this phenomenon. To investigate the role of ER stress in macrophage polarization, the researchers used a culture system in which bone marrow-derived macrophages were exposed to tumor cell-conditioned media. This induced lipid accumulation in the macrophages and skewing to the M2 phenotype, suggesting tumor cells produce M2-inducing factors. The cells produced IRE1-induced fully spliced XBP1, a key factor involved in ER stress. To target this signaling pathway, an IRE1 inhibitor was added to the coculture, which inhibited the skewing to M2 macrophages and their suppression of CD8+ T cell expansion in vitro. In XBP1-knockout mice a lower level of immunosuppressive iTAMs was found and tumor growth was slowed, also suggesting XBP1 induces immunosuppressive TAMs. When lipids were removed from conditioned media, ER stress was not induced in the macrophages. RNAseq data then showed that macrophages upregulate the scavenger receptor mincle after exposure to conditioned media. The mincle receptor recognizes β-glycoceramide, which is highly expressed in the TME. When mincle was blocked by antibodies in the culture system, there was less skewing to M2 phenotype. Furthermore, knockdown of the enzyme controlling β -glycoceramide production resulted in reduced M2 skewing and lower expression of XBP1. Ho and team then showed that tumor conditioned media induced lipid reshuffling on the ER membrane that promotes IRE-1-dependent XBP1 activation. This could be overcome by rewiring of lipid metabolism with LPCAT3 overexpression, resulting in more polyunsaturated fatty acids in the membrane and reduced M2 markers. The mechanism was confirmed in vivo, where an LXR agonist that leads to LPCAT3 activation suppressed the polarization of TAMs and reduced tumor growth in mice.
Tumor microenvironment metabolism in T cell differentiation and dysfunction- Greg M. Delgoffe - University of Pittsburgh, Pittsburgh, PA
The demands of tumor growth set up a highly challenging metabolic state within the tumor microenvironment (TME,) as key nutrients are reduced and toxic by-products build up. Greg Delgoffe asked how immune responses, which are also metabolically demanding, are shaped by this environment, with a focus on tumor infiltrating lymphocytes (TILs). Analysis of T cells over both space (distal lymph nodes, draining LN and intratumoral) and time indicate a progressive track toward metabolic insufficiency (decreased glucose uptake and decreased mitochondrial function) and toward enhanced inhibitory co-receptor (PD-1, LAG-3, etc.) expression, driven by decreased expression of PCG1α, a transcriptional co-activator of mitochondrial production. Overexpression of PCG1α in T cells reversed these effects. Delgoffe then asked whether the march of TILs toward exhaustion was metabolically driven and showed that hypoxic conditions along with chronic TCR stimulation recapitulated the appearance of classic exhaustion markers (PD-1, LAG-3, TOX, CD39) and loss of functionality. A deeper dive based on RNAseq uncovered a critical role for mitochondrial reactive oxygen species (ROS). Specifically increasing ROS in mitochondria with antimycin-A drove exhaustion of T cells and this effect was reversible with a second drug that shut down the respiratory channel and all ROS production. In vivo treatment of mice with axitinib, an inhibitor of VEGFR which normalizes the vasculature and reduces hypoxia, led to better effector function of intratumoral T cells and tumor control, particularly in concert with immune checkpoint blockade (ICB). This observation has led to an ongoing clinical trial of the combination. Delgoffe closed by asking the novel question - what is the function of exhausted T cells? Terminally exhausted CD8+ T cells (Texh) and regulatory T cells (Tregs) from mice share many co-inhibitory receptors (CTLA-4, LAG-3), metabolic (glucose uptake, reduced mitochondrial function, hypoxia exposure), and regulatory (Helios, Nrp1, CD25) features, and are more abundant than Tregs in B16-F10 tumors, prompting the analysis of whether they share immunosuppressive capabilities. Surprisingly, Texh were equally suppressive to Tregs in conventional assays used to analyze Treg suppression. Mechanistically, suppression was not due to IL-10 nor cytotoxicity but to adenosine production, and depended on CD39 expression by the Texh cells. Deletion of CD39 led to loss of suppressive capacity. CD73, needed to complete the conversion of ATP to adenosine, is missing from Texh cells but is supplied in trans from other cells, presumably antigen-presenting cells (stimulation of Texh with co-stimulatory beads did not result in the suppressive phenotype). Finally, Delgoffe closed by showing that targeted deletion of CD39 in CD8 cells in vivo improved tumor control and synergized with ICB.
Chimeric antigen receptor macrophages for the treatment of solid tumors- Saar Gill - University of Pennsylvania, Philadelphia, PA
In an attempt to overcome three well-established barriers to CAR T cell therapy for solid tumors – limited trafficking to and penetration of tumors; a poorly supportive tumor microenvironment; and single target antigen heterogeneity - Saar Gill turned to myeloid cells, the most abundant type of cell within most tumors, and to macrophages in particular. Tumors have evolved to recruit macrophages, which, as tumor-associated macrophage (TAMs), exert many pro-tumorigenic functions. Gill envisioned CAR-bearing M1-type macrophages (CAR-M) as Trojan horses that, following recruitment by the tumor, could both activate innate immunity and, due to antigen processing capabilities, create a vaccinal effect and present other phagocytosed intracellular antigens to T cells. Adenovirus 5 was used to efficiently deliver HER2-targeted CAR constructs to PBMC-derived M1-like macrophages, which were shown to be more effective in phagocytosis and tumor cell killing in vitro than the well-described anti-HER2 antibody trastuzumab, and improved survival in a xenograft model. These beneficial effects were due to both soluble factors from CAR-M that converted bystander M2-type TAMs to a pro-inflammatory M1 phenotype, and enhanced recruitment of and antigen presentation to both resting and activated T cells. In an immunocompetent HER2 mouse model, CAR-M enhanced survival, increased T cell number and activity in tumors and generated/expanded additional tumor-specific T cells that provided complete control to rechallenge with non-HER2-expressing parental tumor cells. These effects were enhanced with anti-PD-1 therapy. A partially-automated 7-day manufacturing process that could yield billions of CAR-M from CD14+ monocytes was developed, and a phase 1 clinical trial in HER2-positive solid tumors is underway.
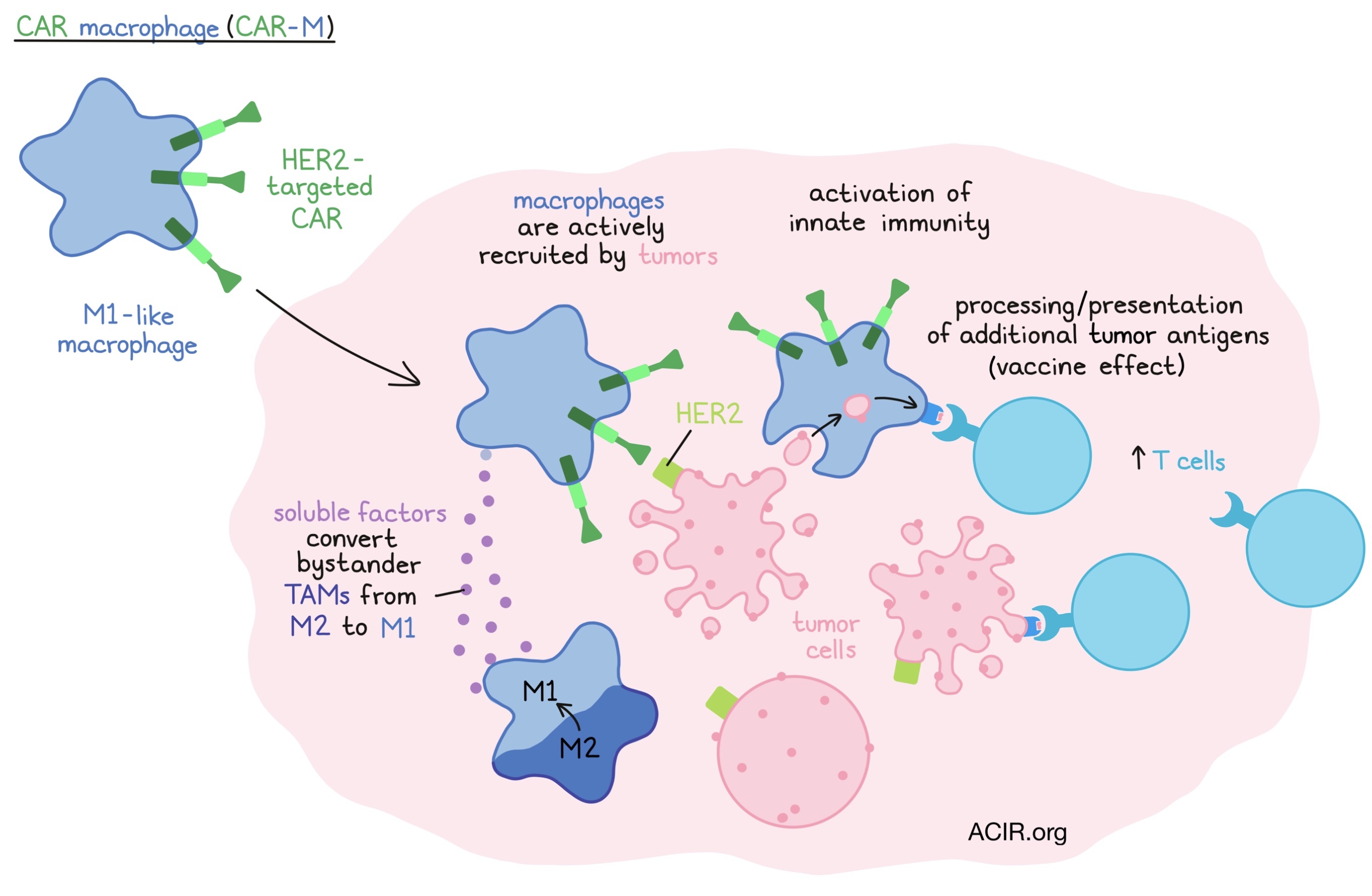
Exploring and therapeutically targeting the tumor microenvironment in brain cancers- Johanna A. Joyce - University of Lausanne, Lausanne, Switzerland
The tumor microenvironment (TME) evolves dynamically during cancer progression and in response to therapeutic intervention, and has organ-specific differences, making it challenging to target the TME. The brain is a unique organ with many cell types that can only be found within the brain. Johanna Joyce and her team used an integrated pipeline to analyze over 300 resected brain tumor and matched blood samples from patients who were diagnosed with brain metastases (originating from melanoma, lung, or breast cancer) or low-grade or high-grade glioma, and compared these samples with non-malignant brain and healthy donor blood samples. The researchers found that the TME immune cell landscape was shaped in a disease-specific manner, and that even the TME of metastases originating from distinctive primary tumor types differed in terms of relative immune composition. Taking a closer look at tumor-associated macrophages (TAMs), which collectively comprise about 25-80% of immune cells across these brain malignancies, Joyce et al. identified cell type-specific and disease-specific patterns of TAM education based on gene expression. In brain metastases, resident microglia and recruited monocyte-derived macrophages are also spatially poised to interact with T cells and exhibit a more immunomodulatory phenotype than in primary brain tumors. In the second part of her talk, Joyce explored whether TAMs are an actionable therapeutic target and whether CSF-1R inhibitors show efficacy across different brain tumors. Joyce and her team have previously shown the small molecule CSF-1R inhibitor BLZ945 (Novartis) to be potent in the treatment of glioma. Contrary to expectations, TAMs in glioma survived despite CSF-1R inhibition, and instead were ‘reeducated’ with increased phagocytosis of tumor cells by the TAMs. The researchers also demonstrated in a prophylactic study that brain-resident microglia are actively involved in the initial steps of metastatic colonization of the brain and that CSF-1R inhibition regressed established brain metastases in preclinical models, but this effect was transitory. The adaptive resistance response to CSF-1R inhibition was accompanied by induction of inflammatory pathways and enhanced CSF2/STAT5 signaling in the protective perivascular niche, leading to survival of an inflammatory TAM subset. This tumor regrowth in protective niches enriched with inflammatory TAMs can be viewed as a consequence of an attempt to repair neurons that have been damaged by the released cytokines. Combination of BLZ945 with a STAT5 inhibitor reduced tumor regrowth and combinations of CSF-1R inhibitors and anti-PD-1 are currently being tested.
Targeting tumor associated macrophages for cancer therapy- Jennifer L. Guerriero - Brigham and Women’s Hospital, Boston, MA
Intratumoral macrophages represent a major proportion of the cells in most tumors, typically get converted to one of multiple pro-tumorigenic phenotypes, and are prognostic of poor relapse-free and overall survival. Multiple approaches to target these suppressive cells are under active investigation. BRCA1-mutated, triple-negative breast cancer (TNBC) is a challenging tumor type, and although approved PARP inhibitors slow tumor growth, recurrence is almost universal. The observation that the efficacy of PARP inhibitors depended on induction of the cGAS/STING pathway and recruitment of CD8+ T cells encouraged the early clinical testing of combination with checkpoint blockade, but without benefit. The high infiltration of suppressive macrophages in BRCA1-mutated TNBC led Jennifer Guerriero to hypothesize that suppressive macrophages recruited by PARP inhibitors were preventing durable responses. After confirming the enhanced infiltration of both CD8+ T cells and macrophages in BRCA1-mutated TNBC using multi-targeted cyclic immunofluorescence, Guerriero and her team turned to a GEMM TNBC model that recapitulated the enhanced CD8+ T cell and macrophage infiltration, and demonstrated significant induction of the cGAS/STING pathway, along with increased CD80, PD-L1 and CSF-1R (the CSF-1 [‘macrophage CSF’] receptor) on the macrophages. To determine if PARP inhibitors had a direct effect on macrophage differentiation, CD14+ human monocytes were differentiated with IL-4 and GM-CSF ex vivo. Co-treatment with PARP inhibitors caused similar phenotypic changes. Hypothesizing that the combination of PARP inhibitor and blockade of CSF-1R would be more effective, Guerriero and her team turned to the GEMM model and demonstrated that the combination significantly improved survival, which depended on CD8+ T cells, but animals still relapsed. Mechanistically, in vitro-generated human macrophages treated with PARP inhibitors released soluble factors that suppressed T cells and led to increased T cell apoptosis. RNA sequencing, proteomic analysis, and metabolic assays demonstrated a shift from glycolytic to lipid metabolism of the treated macrophages. Interestingly, PARP inhibitors reduced glucose uptake in macrophages, consistent with a shift to lipid metabolism, but this reduction was independent of PARP (using PARP KOs), suggesting an alternate pathway. This effect could be explained by recent results from the Vignali lab indicating that the SREBP1-mediated fatty acid synthesis pathway is associated with pro-tumor macrophages. Guerriero et al. showed that inhibitors of SREBP1 could inhibit this PARP inhibitor-induced macrophage conversion in vitro, but surprisingly SREBP1 inhibition did not enhance the effect of PARP inhibition in vivo. Fortunately, the triple combination of PARP inhibitor (which killed tumor cells and induced cGAS/STING and CD8+ T cell infiltration, but also induced the release of CSF1 and recruitment of suppressive macrophages), SREBP1 inhibitor (which blocked the metabolic conversion), and anti-CSF-1R (which further down-modulated the suppressive macrophages) resulted in impressive long-term control in the GEMM model.
Back To Top
Tumor immunity
Mapping myeloid programs that control tumor immunity- Miriam Merad - Icahn School of Medicine at Mount Sinai, New York, NY
In her Keynote Lecture, Miriam Merad described her goal of building a better understanding of antigen-presenting cells, which are critical to the T cell stimulation that underlies checkpoint blockade. Acknowledging a potentially important role for inflammatory monocyte-derived dendritic cells (mo-DCs) of the macrophage lineage, her talk focused on cells of the dendritic cell lineage derived from bone marrow CMP/CDP precursors, which appear to have evolved resistance to viral infection and so developed cross-presentation capability as a means to internalize antigens from other infected cells for presentation to T cells. Combining CITE-Seq data across multiple early, treatment-naive tumors from lung, liver, and colorectal cancer, with unbiased analysis generated expression programs that were compatible with other published data sets and revealed a commonality of patterns across patients and tumor types. Focusing the analysis on antigen-presenting dendritic cells revealed not only the canonical cDC1, cDC2, and moDCs, and the relationships between them (with moDCs being closer to CD14+ monocytes and cDC2s, and cDC1s being more distinct), but also an additional class of DCs found in all tumor types and with analogs in the mouse. These ‘cDCs’ expressed strong signs of maturation and activation (CD40, CD80/86), but also strongly expressed regulatory modules (PD-L1, PD-L2, CD200, etc.) and were termed mregDCs (mature DCs enriched in immunoregulatory markers). These are equivalent to separately described DC3s and LAMP-3 DCs. Additionally, mregDCs were high in migration-related gene expression and low in TLR gene expression, apparently limited in environmental “sensing”. Both cDC1s and cDC2s have mregDC equivalents, indicating that the mregDC program identifies a distinct differentiation state of DCs rather than a new class. In vivo studies showed that tumor antigen-charged DCs were enriched in the mregDC program, and in vitro studies showed that antigen uptake induced cDC1 or cDC2 conversion to the mregDC program. Moreover, using a technique to isolate interacting cells from tumor samples (PIC-seq), mregDCs were the predominant APC type interacting with T cells in tumors, and these cells were found predominantly in tertiary lymphoid structures, which are known to actively recruit T cells. These results suggest that mregDCs, which show both activation and negative regulatory potential, may be the predominant APC type interacting with naive T cells, and so finding ways to uncouple their immunogenic versus tolerogenic programs may be beneficial. Mechanistic studies showed that IL-12 is critical for the Th1 response, but this can be abrogated by IL-4, and that IL-4R blockade promotes tumor regression by enhancing IL-12 production of tumor antigen-bearing mregDCs. Merad and her team are now investigating the benefit of combination IL-4R/PD-1 blockade in a clinical trial in patients with metastatic lung cancer. Attempting to discover new targets that can affect the stimulatory/regulatory balance in mregDCs, Merad described a novel CRISPR screen, which identified SOCS1 as an important positive regulator for the mregDC program, opening up SOCS1 and its regulators as novel targets.
The immune contexture and its impact on cancer treatment- Jérôme Galon - INSERM, Paris, France
Jérôme Galon presented the multiple uses of the tumor immune contexture to evaluate and improve therapy response in cancer. The immune contexture can be measured in two ways; the Immunocore, which takes into account the type, density, and location of immune cells as determined by IHC (quantitative), and the Immunosign, which uses gene expression profiling to define qualitative immune signatures. Such data can be used to generate both predictive and prognostic markers as well as give mechanistic insights. In rectum cancer, the response to neoadjuvant radiochemotherapy could be predicted by the Immunoscore of biopsy samples. A high score was correlated with no relapses, suggesting it might be a good indicator for patients who would benefit from a watch and wait strategy (radiochemotherapy but no surgery). This was validated in a study following patients undergoing the watch and wait approach; those patients with a high immunoscore score did not have any relapses. Therefore, the Immunoscore of the biopsy can predict clinical outcome, identify patients eligible for watch and wait, and might be used to restage local disease after neoadjuvant treatment. Galon and colleagues also assessed the use of the immune contexture to predict response to CAR-T therapy. CD19-CAR-T is an approved therapy for patients with diffuse large B-cell lymphoma (DLBCL) with relapsed or refractory disease after ≥2 lines of systemic therapy. Studying patients with DLBCL before and after being treated with CD19-CAR-T, the researchers found major changes in many immune-related genes post-treatment, such as checkpoint, IFN-related, effector, proliferation, and chemokine genes. The pre-treatment Immunosign score could separate responders from non-responders, and 80% of the complete responders had a high Immunosign score. In addition, the Immunoscore and Immunosign were both associated with prolonged survival and, along with baseline tumor burden, were the only predictive factors in multivariate analyses. In particular, this data revealed a potential mechanistic link between myeloid cell-derived cytokines and T cell phenotype and that high densities of CD3+CD8+PD-1+LAG3+/-Tim3- T cells in pretreatment biopsies were associated with response to CAR-T therapy.
Lymph node colonization promotes distant tumor metastasis through the induction of tumor-specific immune tolerance- Nathan Reticker-Flynn - Stanford University, Menlo Park, CA
The vast majority of solid tumors first spread to lymph nodes (LN), and lymph node involvement represents a determinant of worse prognosis across cancers. To understand the role of lymph node metastasis in tumor progression, Nathan Reticker-Flynn and colleagues generated a panel of tumor cell lines with enhanced capacity to metastasize to lymph nodes, starting with non-metastatic B16-F0. Later-generation cell lines with high capacity for metastasizing to lymph nodes rendered syngeneic mice susceptible to distant organ metastases, confirming that lymph node tumors promote metastatic seeding of distant sites. RNAseq and ATACseq analysis of the various LN tumor cell lines revealed the gradual acquisition of a distinct transcriptional signature dominated by Type I and II interferon response genes (IRG), increased chromatin accessibility at IRG loci in later-generation LN cell lines, and increased STAT transcription factor activity. IFNR expression and continual IFN stimulation were required for the IRG signature and epigenetic reprogramming of the LN metastases. Tumor colonization of the LN also induced a broad constellation of changes in the immune repertoire, with B cell alteration towards an IFN-dominated state, changes in CD8+ and CD4+ T cell clusters from naive to activated and/or memory subsets, increases in macrophage and resident DC populations, and decreases in migratory DC numbers. Most dominant though was an increase in Tregs. Tregs from mice bearing LN tumors incubated with naive CD4+ T cells induced the conversion of naive CD4+ T cells to Tregs at a much higher rate than when Tregs from tumor-free mice were used. Mice formed fewer lung metastases in the absence of Tregs, and tracking experiments revealed that Tregs from mice with LN metastases traveled to the lungs at a much higher rate than Tregs from mice without LN metastases. Reticker-Flynn et al. used different mouse models to learn if the TCR specificity of Tregs played a role in the enhanced metastases formation, and observed that LN metastases enhanced the induction of antigen-specific Tregs, and that these antigen-specific Tregs are likely to facilitate the metastatic seeding of distant sites. These findings may translate to human data, as HNSCC patients with LN metastases had greater numbers of Tregs and an IRG-dominated transcriptional signature in their affected LNs.
Quiescent cancer cells form immunotherapy resistant reservoirs by forming an immune suppressive niche- Judith Agudo - Dana-Farber Cancer Institute, Boston, MA
When treating mice bearing GFP-positive triple-negative breast cancer (TNBC) with GFP-specific CD8+ T cells (Jedi T cell) Judith Agudo and colleagues found that this treatment was able to shrink, but not fully eliminate tumors. Treatment-resistant, but antigen-expressing cancer cells were still present, and these cells had downregulated gene sets related to proliferation, did not incorporate the thymidine analog Edu into their DNA, and were identified as quiescent using the mVenus-p27k ‘quiescence’ reporter described by Oki et al. (2014). The quiescent cells showed a cancer stem cell phenotype and, thus, higher tumorigenic potential than their proliferative counterparts. Fluorescent histology of tumor tissue revealed clusters of quiescent cancer cells that excluded infiltrating T cells. To characterize the quiescent cancer cell niches, Agudo and her team developed a technique called PADME-seq (photo-conversion of areas to dissect micro-environments). TNBC tumor cells expressing a tomato-p27k quiescence reporter were transplanted to transgenic mice expressing the photoconvertible fluorescent protein Kaede, which changes under UV light from green to yellow. Areas of tumor sections containing the quiescent cells could be identified under a fluorescence microscope by the tomato red color, allowing marking of areas containing quiescent tumor cells (red) or non-quiescent tumor cells (no color). Infiltrating cells from the Kaede+ host are identified as green. Marked areas could then precisely be exposed to UV light and photoconverted, which labels tumor-infiltrating host cells in that area yellow, enabling flow sorting of infiltrating cells from each region of interest for single-cell sequencing. Tumor-infiltrating T cells were less abundant in quiescent cancer cell clusters, and a higher percentage of these T cells expressed high levels of exhaustion markers. Furthermore, fibroblasts and DCs isolated from the quiescent cancer cell regions were shown to be dysfunctional and immunosuppressive.
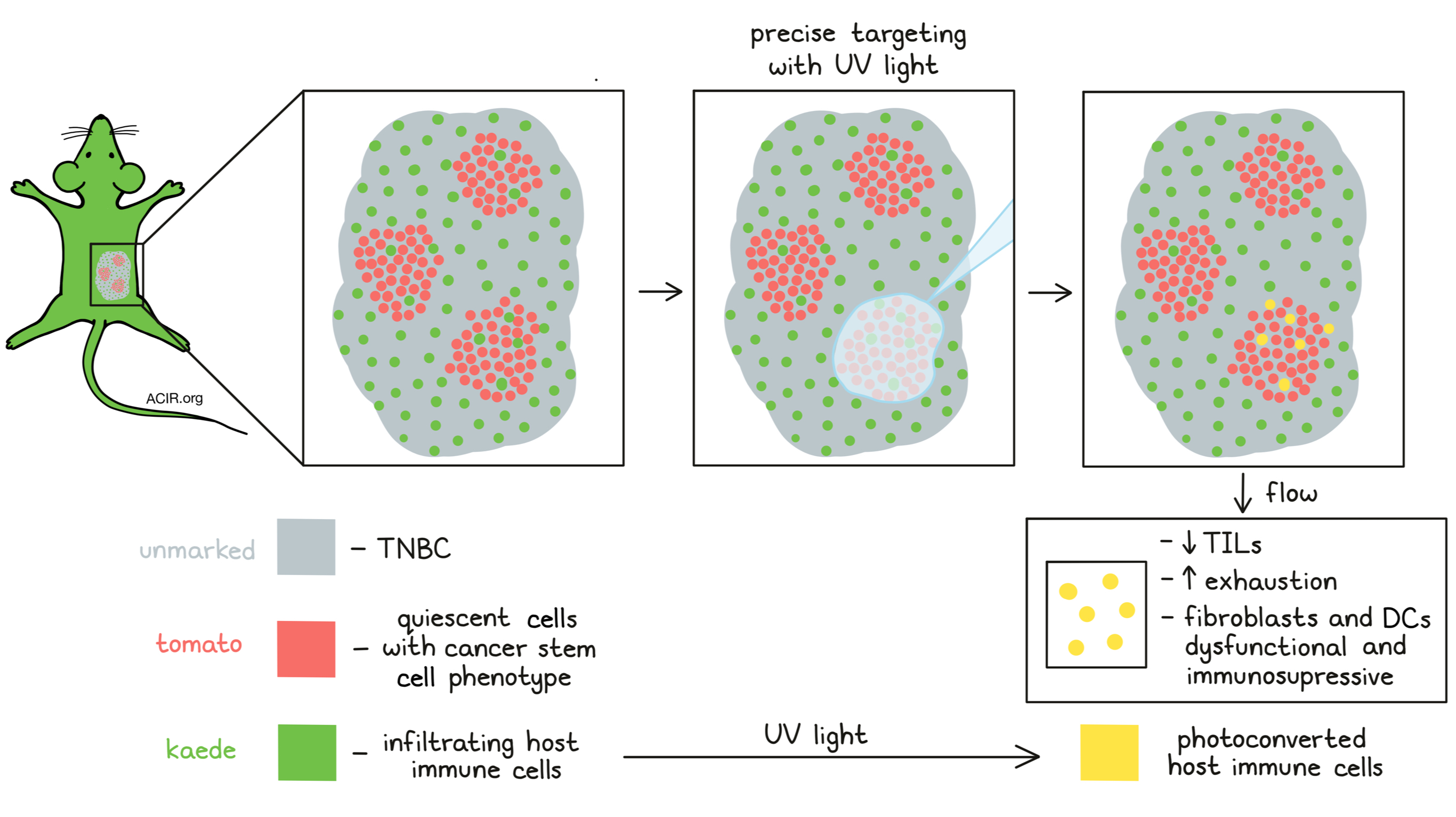
T cell therapies
Engineering enhanced persistence and anti-tumor function for therapeutic human T cells- Caroline Arber - Lausanne University Hospital, Epalinges, Switzerland
Caroline Arber presented strategies to improve adoptive TCR T cell therapies. Besides TCR antigen recognition, costimulation and cytokine support are also necessary to activate T cells. These latter signals are often lacking in the tumor microenvironment (TME). To improve therapy efficacy, Arber’s group developed two strategies using a transgenic TCRαβ that targets the tumor antigen survivin, which is overexpressed in many hematological and solid tumors, in context of HLA-A*02:01. The first strategy exploited the CD8αβ co-receptor function. Transgenic expression of the survivin-specific TCR and the CD8 coreceptor (TCR8) in CD4+ and CD8+ T cells resulted in effective in vitro and in vivo tumor cell killing in a MHC class I-restricted manner. scRNAseq revealed that inclusion of the CD8αβ transgene increased the transcription of naïve or central memory programs in both TCR8+CD4+ and TCR8+CD8+ T cells. The CD4+ subset differentially expressed genes related to cytotoxicity, co-stimulation, oxidative phosphorylation (OXPHOS), and proliferation, while exhaustion genes were reduced. On the other hand, the TCR8+CD8+ subset was enriched for exhaustion genes, and had diminished OXPHOS and proliferation gene expression. In conclusion, TCR8+CD4+ demonstrated the highest potential for the development of enhanced TCR T cell therapies, outperforming TCR8+CD8+ T cells. The second strategy was focused on delivering costimulatory and cytokine signals to adoptively transferred TCR T cells in the TME. Arber and her team engineered TCR T cells to express c-MPL, the receptor for thrombopoietin (TPO). TPO is present in the bone marrow TME of hematological malignancies and FDA-approved pharmaceutical c-MPL agonists are also available. Signaling through c-MPL activates pathways shared with common T cell costimulatory and γ-chain cytokine receptor signaling. c-MPL+ TCR-engineered T cells demonstrated enhanced expansion and anti-tumor activity in vitro and in vivo in the presence of TPO or a c-MPL agonist. c-MPL+ TCR T cells preserved a central memory phenotype and c-MPL signaling resulted in more efficient immune synapse formation and activation of type I IFN pathways.
Next generation CAR T cells to overcome resistance- Crystal L. Mackall - Stanford University, Stanford, CA
Building on the successful approval of CAR therapies across multiple hematologic disease settings, a dramatically increasing number of CAR T cell clinical trials are in progress, highlighting the need to assess CAR T cell performance with respect to the important goal of long-term progression-free outcomes. In her Keynote lecture, Crystal Mackall discussed the research into overcoming one of the main mechanisms behind therapy resistance – tumor heterogeneity. In B-cell acute lymphoblastic leukemia (B-ALL), half of the relapses after CD19-CAR-T therapy are CD19 negative. Currently, the tools to identify patients a priori who are at increased risk of these relapses are lacking. To assess whether CD19 loss indeed was responsible for the relapses, Mackall reviewed the data from a trial with a CD22-CAR-T which included 57% of patients who had CD19neg/dim expression, most of whom had been previously treated with a CD19-CAR. 70% of patients showed complete responses, including the CD19neg/dim subset. Disappointingly, the relapse-free survival was only 6 months and although there was occasional loss of CD22, more commonly relapsers showed a lower level of CD22 expression. This led to a focus on antigen density, consistent with prior findings that a distinguishing feature between CARs and natural TCRs is that CAR T cells require recognition of thousands of antigens to get activated, while TCR T cells require only 10-100 molecules. This antigen density requirement is impacted by the costimulatory domain, the hinge, T cell exhaustion, scFv affinity, and CAR receptor expression density. To overcome this resistance mechanism, simultaneous targeting of two antigens by CAR T cells was investigated. In a phase I study a loop CAR with an OR gate for CD19 and CD22 scFvs was tested in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and B-ALL. This treatment turned out to not add toxicity to the single CAR-T profile, and the response rates in both DLBCL and B-ALL were similar to those of the CD19 single CAR T cells, but still with high rate of relapses. In these patients, there was again a loss of CD19 expression, but not CD22. This led Mackall and her team to hypothesize that the immune pressure on CD22 mediated by this dual CAR T cell was inadequate to prevent CD19-/neg relapses. They confirmed this using engineered cell lines and single cell analysis platforms, showing that the dual CAR T cells manifested higher activation to the CD19 than the CD22 antigen. When comparing the potency of the dual CAR T cells with the two single CAR T cells, they found that the monospecific CD22 CAR T cells exhibited greater potency to CD22 than the bispecific CAR T cells, with comparable production of IFNγ and upregulation of CD69 and CD107a but reduced production of TNFα and IL-2. These data suggest that TNFα and IL-2 are more important to assess as markers of potency than IFNγ or killing activity. Therefore, a remaining challenge is to ensure an equivalent potency across multiple antigens in multi-specific CAR constructs. Mackall closed by describing remarkable results of a clinical trial with a CD22-CAR-T in large B cell lymphoma, demonstrating good safety and 60% durable progression-free survival in patients who had progressed after multiple prior therapies, including CD19-CAR-T therapy.
Back to Top
Systems Biology
Mapping the dynamics of immunotherapy to quantify tumor antigenicity- Grégoire Altan-Bonnet - National Cancer Institute, Bethesda, MD
With the aim of producing more consistent and more effective tumor-infiltrating lymphocyte (TIL) products, Grégoire Altan-Bonnet and colleagues used CYTOF with a panel of 38 markers to profile TIL products from multiple patients.They found a high phenotypic diversity, both between and within patients, even though all TILs were proliferated under the same culture conditions. Deeper analysis of samples from 34 patients revealed that the phenotype could predict the outcome of TIL therapy, as responders had increased frequencies of CD39-/lowCD69-/low (stem cell-like) T cells. This was confirmed in a B16 mouse model, where these less exhausted/dysfunctional stem-like TILs mediated anti-tumor activity. Single cell RNAseq analysis of 11 patients tumor samples immediately following surgical revision revealed a large diversity of T cell phenotypes, but interestingly specific clonotypes yielded similar phenotypes. To assess in more detail how many types of TILs exist and what the best subtypes are for adoptive TIL therapy, Altan-Bonnet and colleagues developed an ex vivo model for T cell activation. For discrimination between self and non-self-antigens, T cells need to differentiate peptide quality - the binding affinity of the antigen plus MHC to the TCR - and quantity - the antigen density at the surface of antigen-presenting cells. To model TCR signaling, antigen-recognition of specific peptides and cytokine production need to be considered. The researchers used a custom-built robotic platform to measure immune dynamics of T cell activation, allowing highly multiplex measurements of 23 cytokines and 15 other markers across 18 different time points in response to different antigens. This analysis produced a rich data set with more than 10,000 observations and this complex raw data required de-convolution with dimensionality reduction and neural network-based machine learning to obtain a simple representation. Their model estimated that at least six distinct classes of antigen exist across a range of effectiveness. This information can now be used for multiple applications in which mouse T cell data is used as the training set and smaller human clinical sample sets (TILs or CAR-T) as the test set to map the T cells responding to tumors.
Back to Top
Combination therapy
Understanding responses to cancer therapy: The tissue is the issue, the scoop is in the poop, and you are what you eat- Jennifer A. Wargo - The University of Texas MD Anderson Cancer Center, Houston, TX
As summarized by Jennifer Wargo, two of the main current challenges in the immunotherapy field are predicting therapy response and identifying factors that alter the response. Timing is important, and as immunotherapy is being used in earlier stages of treatment, the efficacy has increased. In particular, when patients who would frequently relapse after surgery are now first treated with neoadjuvant immune checkpoint blockade (ICB), they are significantly less likely to relapse, particularly if they showed a pathological complete response. In comparison, neoadjuvant TKI-targeted therapy can also be initially effective, but long-term responses are not as reliable as those induced by ICB. In melanoma, most patients benefit from neoadjuvant ICB, and the use of neoadjuvant ICB is expanding to multiple cancer types. Tumor mutation burden and an IFNγ signature have emerged as predictive biomarkers. Wargo next outlined multiple host intrinsic and extrinsic factors that impact immunity and should be taken into account when designing rational combination strategies to improve outcomes. The host microbiome has been shown to affect therapy responses to ICB. Intratumoral microbes may impact anti-tumor immunity; however, the interactions are complex. In some cases, intratumoral microbes are associated with a more suppressive tumor microenvironment, while more diverse microbiomes in a tumor are associated with improved outcome and increased immune infiltration. There is also a complex and dynamic interplay between microbes and immune cells in the gut. Early studies showed that the diversity and composition of the gut microbiome could shape the response to immunotherapy, although again, the devil is in the details. Broad antibiotic treatments can reduce anti-tumor immunity, while drugs targeted to individual species may improve anti-tumor immunity. Emerging evidence points to an important role for the induction of tertiary lymphoid structures and B cell responses, and a current clinical trial is assessing the impact of perioperative antibiotics on the gut microbiota and clinical outcomes. Diet may also play a role, as patients with sufficient fiber intake have better outcomes to ICB. Fiber intake was shown in preclinical mouse models to shape the tumor microenvironment, resulting in an increase in dendritic cell infiltration, a delay in tumor outgrowth, and monocyte reprogramming through STING-mediated IFN signaling. On the other hand, the use of commercially available probiotics negatively impacts therapy response. Therefore, dietary intervention trials are currently being run in combination with ICB to assess its impact on efficacy more rigorously.
Immunotherapy for melanoma: Checkpoint blockade combinations- Jedd D. Wolchok - Memorial Sloan Kettering Cancer Center, New York, NY
Jedd Wolchock gave an update on checkpoint blockade therapy for melanoma, especially around the use of combinations to improve outcomes. The anti-CTLA-4 antibody ipilimumab (IPI) was the first checkpoint blockade therapy to be approved. Blocking CTLA-4 in the context of cancer can lead to a super-physiological immune cell infiltration (macrophages and lymphocytes) into the tumor mass. Over 20% of patients with metastatic melanoma have a long-lasting survival benefit with 5 years or longer from treatment with IPI compared to a 2-5% 5-year survival before the approval of IPI. Side effects of IPI resemble autoimmune disease with specificity to self-antigens present in the skin, mucosa of the GI tract, and parenchymal cells of the liver and endocrine organs, and, if detected early, in most cases are reversible (with exception of endocrinopathies which often require permanent hormone replacement therapies). Various antibodies blocking PD-1 or PD-L1 were the next checkpoint blockade inhibitors to be approved. Adverse events were less frequent with these antibodies and no major differences in biological activity have been observed between receptor or ligand blocking antibodies. Early studies revealed no cross-resistance to CTLA-4 and PD-1/PD-L1 checkpoint blockade inhibitors, which led to combination studies. Wolchock summarized results from the CheckMate 067 phase 3 study that were presented at ASCO this year. 945 patients with newly diagnosed metastatic melanoma were either treated with the combination of nivolumab (Nivo) and IPI or the monotherapies alone. At a follow-up of 6.5 years, response rates for Nivo + IPI were 58%, 45% for Nivo alone, and 19% for IPI alone. Encouragingly, the median duration of response has not yet been reached for Nivo+IPI or Nivo alone. The median overall survival (OS) for Nivo+IPI was 72 months (the melanoma-specific OS has not yet been reached for this group), 37 months for Nivo (melanoma-specific OS: 59 mo) and 20 months for IPI (melanoma-specific OS: 22 mo). Grade 3-4 adverse events for combination therapy were 59%, more than double compared to 24% and 28% for the monotherapies. Another interesting immune checkpoint is LAG-3. LAG-3 and PD-1 both contribute to T cell exhaustion and are often co-expressed on tumor-infiltrating lymphocytes. Blockade of LAG-3 and PD-1 has revealed synergistic antitumor activity in preclinical models. Wolchock pointed out results from the RELATIVITY-047 trial that were presented by Evan Lipson at ASCO this year. In this phase 2/3 study, 714 patients with previously untreated unresected or metastatic melanoma were treated with the LAG-3 blocking antibody relatlimab (Rela) and Nivo or Nivo alone. The median progression-free survival (PFS) showed an improvement by about 5.5 months for the combination. Treatment-related grade 3-4 side effects were observed in 19% of the patients for the combination and in 10% with Nivo alone. Also at ASCO was presented a study by Rodabe Amaria investigating the neoadjuvant and adjuvant use of Rela and Nivo in patients with resectable clinical stage III melanoma or oligometastatic stage IV melanoma. At a median follow-up of 16.2 months, the complete pathological response rate for the combination therapy was 59% and toxicity was low. At the end of his talk, Wolchock presented results for a translational analysis supporting the use of LAG-3 blockage. Three distinct ‘immunophenotypes’ were distilled out by multiparametric flow cytometry from banked peripheral blood samples that had been obtained from patients who had been treated with checkpoint blockade. The LAG+ immunophenotype (in part defined by the presence of LAG-3+CD8+ T cells) correlated with poorer OS and PFS in patients treated with checkpoint blockade and was independent of PD-L1 expression. In summary, checkpoint blockage may disinhibit a baseline response to achieve regression and antigen spreading, leading to durable disease control in some patients. Next steps for improvements include supplementation of the T cell repertoire with engineered T cells or ex vivo expanded TIL in patients with a low baseline response, and tackling suppressive cells, physical barriers to trafficking, deficient antigen presentation/processing, hostile microenvironment, insufficient co-stimulation, and metabolic challenges (glycolytic tumor microenvironment).
Back to Top
By Ute Burkhardt, Ed Fritsch, Lauren Hitchings, and Maartje Wouters



