Tumors create a hostile microenvironment that disrupts T cell metabolism and effector function and allows for tumor progression, but the specific pathways between the environmental stressors and the disruption of T cell metabolic function are not fully understood. In a letter recently published in Nature, Song et al. investigated this mechanism in the context of ovarian cancer, elucidating the intracellular pathways that lead to metabolic dysfunction in T cells and identifying potential points of intervention.
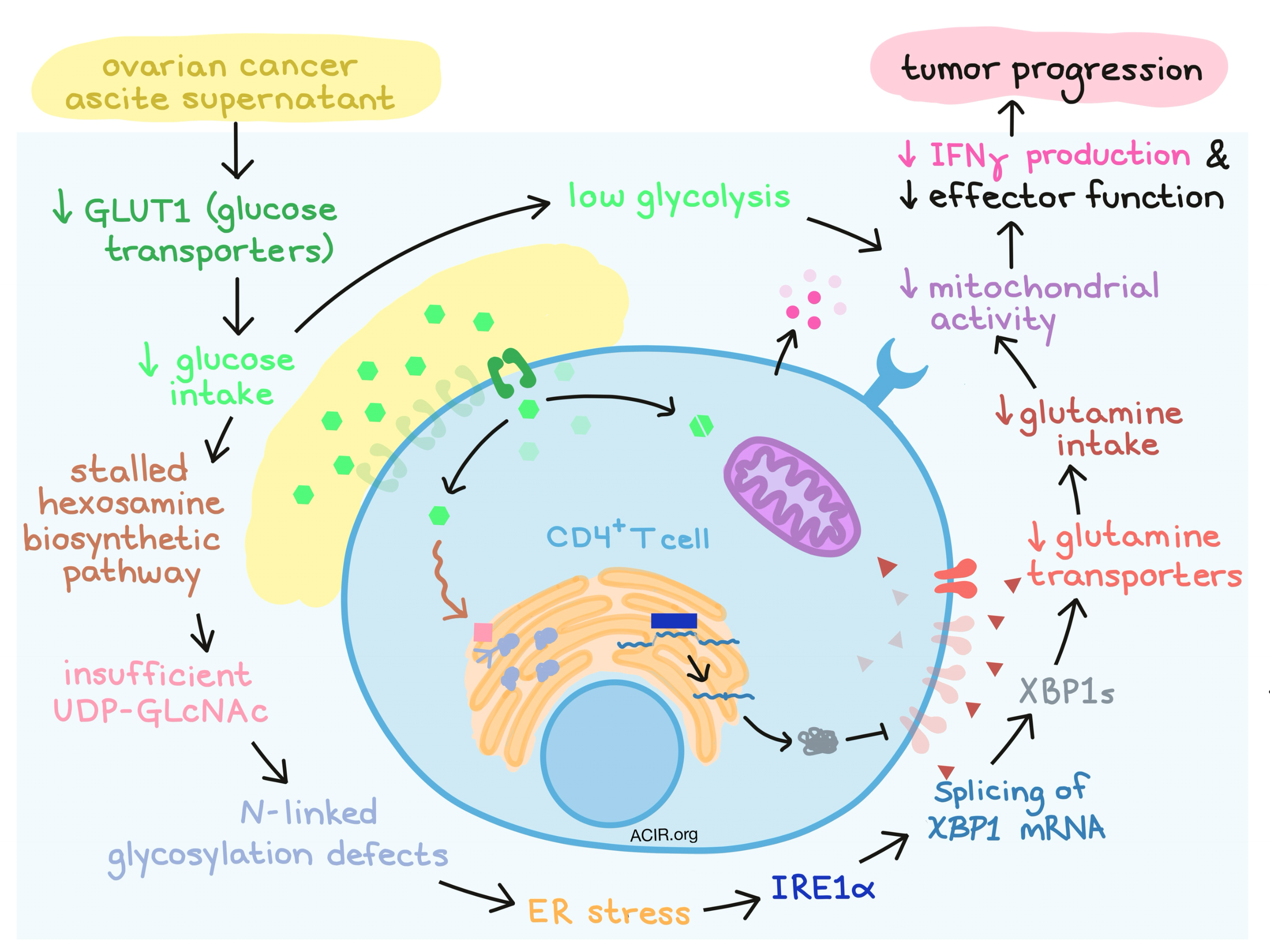
Due to its known role in endoplasmic reticulum (ER) stress, protein folding pathways, and enhanced tumorigenic capacity, Song et al. considered the IRE1α-XBP1 pathway the prime suspect in their investigation. Under conditions of stress in the endoplasmic reticulum (ER), the IRE1α enzyme excises a fragment from XBP1 mRNA, and the spliced version of the mRNA encodes the functionally active form of the XBP1 protein, known as XBP1s. When the researchers isolated CD4+ and CD8+ T cells from patient-derived ovarian cancer ascites, they observed an increase in XBP1 splicing. XBP1 levels also correlated with expression of genes that indicated activation of the unfolded protein response (UPR), and therefore with ER stress. This signature was associated with reduced T cell infiltration, indicating that this pattern may influence T cell functionality.
To better understand the role of IRE1α-XBP1, Song et al. isolated CD4+ T cells from healthy women and found that they increased expression of XBP1 in a dose dependent manner upon exposure to the supernatants of ascites derived from ovarian cancer patients. T cells treated with the ER stressor tunicamycin also showed evidence of strong XBP1 activation that could be abrogated by administering an IRE1α inhibitor, indicating that ER stress induces IRE1α-XBP1.
Working backwards to identify the source of ER stress, the researchers noticed that exposure to ascites fluid suppressed the major glucose transporter GLUT1 in CD4+ T cells, which would compromise glucose uptake and cause internal glucose deprivation. A lack of internal glucose would stall the hexosamine biosynthetic pathway used to make uridine diphosphate N-acetylglucosamine (UDP-GLcNAc), which is required for N-linked protein glycosylation. In line with this established mechanism, ascites-exposed CD4+ T cells showed N-linked protein glycosylation defects associated with ER stress. Supplementing GLcNAc, a direct substrate for N-linked protein glycosylation, minimized ER stress and halted downstream XBP1 activation, validating the link between limited glucose uptake and upregulated IRE1α-XBP1.
Next, Song et al. investigated what happens downstream of XBP1 activation and noticed that the glycolytic capacity and oxygen consumption were low in ascite-exposed CD4+ T cells, implicating mitochondrial dysfunction. Blockade of IRE1α in wild-type cells exposed to ascites fluid abrogated this effect on mitochondrial respiration. Engineered XBP1-deficient CD4+ T cells had superior mitochondrial respiration compared to WT under conditions of glucose withdrawal, indicating a direct role for IRE1α-XBP1 in dampening mitochondrial activity. Mechanistically, the researchers found that XBP1s mediated the activation of an ER-associated degradation system that controls the abundance of glutamine carriers, likely through post-translational mechanisms. A lower number of glutamine carriers limits influx of glutamine, which is needed to sustain mitochondrial respiration when glucose is in limited supply. Treating T cells rendered metabolically dysfunctional by ascites exposure with a glutamine analogue, or forcing overexpression of glutamine transporters, restored some respiratory capacity to the T cells.
The researchers then performed transcriptional analysis on wild-type and XBP1-deficient T cells from mice bearing ovarian tumors and found that within ascites, XBP1-deficient T cells showed global transcriptional reprogramming, upregulation of genes related to activation and mediation of antitumor response, and improved effector capacity compared to wild-type T cells. Mice engineered with XBP1-deficient T cells and challenged with ovarian cancer had a higher portion of CD4+ T cells producing IFNγ and a higher portion of CD8+ T cells with increased IFNγ and perforin production. Further, these mice showed superior antitumor immunity, delayed malignancy, and longer overall survival.
In order to show that their findings in mice were relevant to humans, Song et al. evaluated healthy human CD4+ T cells exposed ex vivo to ascite supernatants derived from ovarian cancer patients and found that IFNγ production, indicative of effector function, decreased. Administration of an IRE1α inhibitor partially alleviated this effect and restored some mitochondrial respiration.
Overall, Song et al. elucidate the pathway by which the ovarian cancer microenvironment reduces mitochondrial activity in CD4+ T cells, allowing for tumor immune escape. What they describe can be considered an “immunometabolic checkpoint” controlling antitumor immunity. In outlining this mechanism, the researchers note several points at which intervention could be applied to restore T cell functionality. Controlling ER stress or inhibiting IRE1α-XBP1 might limit metabolic stress and enhance the anticancer immune response, but further research will be needed to understand the potential benefits and consequences of these strategies.
by Lauren Hitchings



