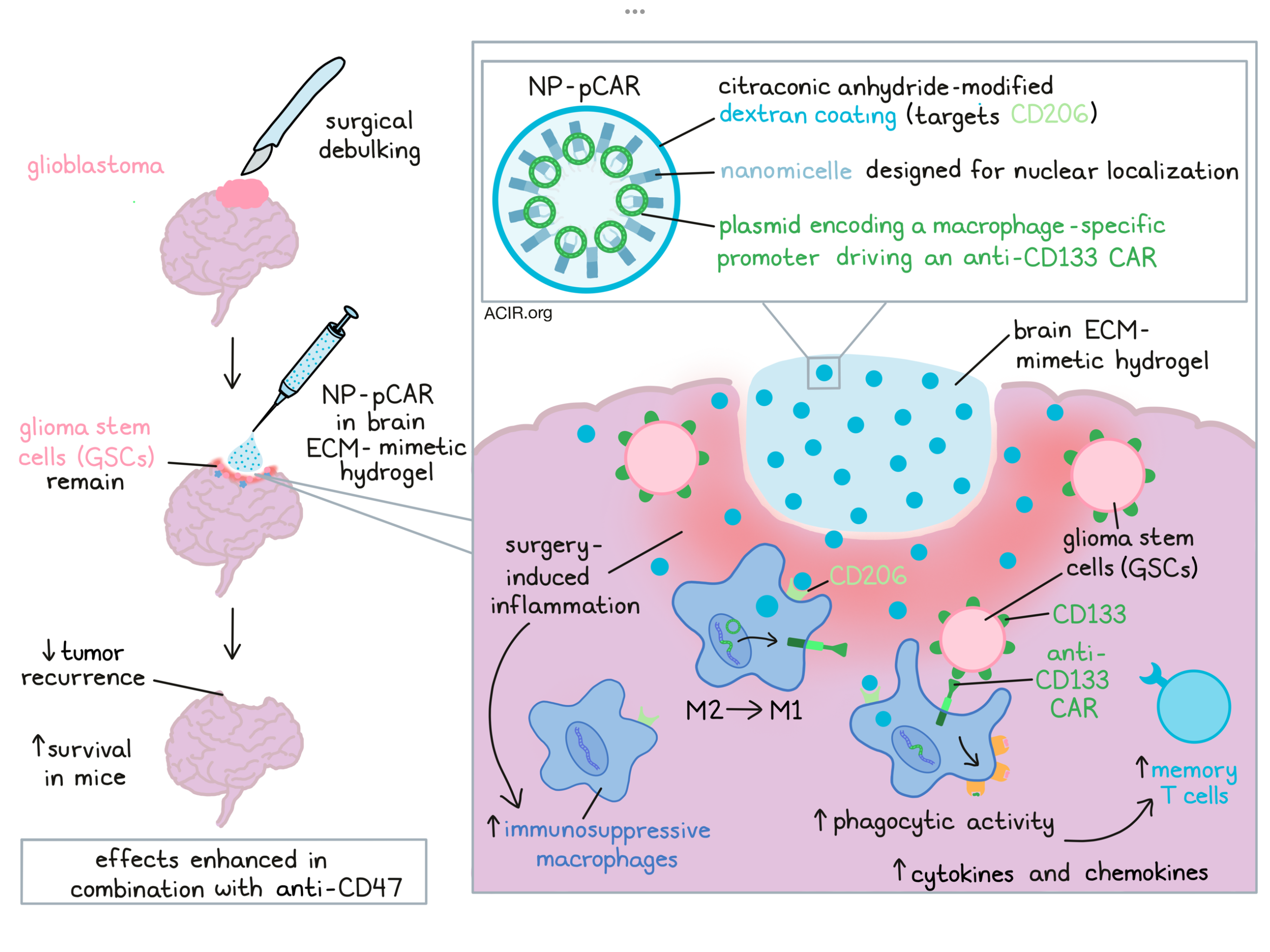
Despite aggressive treatment involving debulking surgery and chemoradiotherapy, glioblastoma multiforme (GBM) remains incurable. A major culprit for GBM recurrence and therapy resistance are self-renewing glioma stem cells (GSCs) that are left behind after surgery. Additionally, immunosuppressive macrophages are recruited to the tumor cavity in response to surgery-induced inflammation. Chen and Jing et al. investigated whether these intracavitary macrophages could be redirected to induce phagocytic activity against GSCs by engineering them to express GSC-specific chimeric antigen receptors (CARs). Their data describing the efficacy of this approach in animal models were recently published in Science Translational Medicine.
Using bioinformatics, the researchers found that CD133 expression was high in glioma tissues, and variable expression levels were detected on patient-derived glioblastoma cells. High CD133 expression was correlated with tumor recurrence and poor survival. Therefore, the researchers constructed macrophage-specific promoter-driven anti-CD133 CAR plasmids (pCAR) encoding the CD3ζ intracellular costimulatory domain. Macrophage-targeted pCAR-laden nanoporters (NP-pCAR) were synthesized with cationic peptides and a brain extracellular matrix (ECM)-mimetic hydrogel was created to serve as the transporter for NP intracavitary immobilization. The NP-pCAR released from the hydrogel was designed to target the macrophages in the cavity and introduce the CD133-specific CAR into the macrophages' nuclei.
The researchers designed a positively amphipathic chimera for nuclear-targeted gene delivery using a nuclear localization sequence peptide as the hydrophilic moiety and palmitic acid as the hydrophobic part. The amphipathic PA-peptide self-assembled into a nanomicelle, into which the negatively charged pCAR was loaded through electrostatic interactions. The pCAR-laden nanomicelle was then further coated with citraconic anhydride-modified dextran to produce a CD206 receptor-targeted pCAR-laden nanomicelle (NP).
To test the efficiency of the NP-mediated gene delivery, the researchers made use of THP-1 cells, bone marrow-derived macrophages (BMDM), and RAW 264.7 cells. In these macrophages, the cellular integration of genes was enhanced when they were encapsulated in dextran-coated NP in vitro. The NP efficiently delivered the genes into macrophages with the dextran-targeting motif; the NP escaped the lysosome and entered the cytosol. It was also shown that the macrophages induced functional CAR expression and the macrophages had increased phagocytic activity against CD133+ cells.
Proteomics analysis of the CAR-macrophages showed that M1-associated pathways were induced. Several M1 macrophage-associated biomarkers were upregulated, while M2 proteomic markers were inhibited. There was an increase in the expression of CD80, iNOS, TNFα, and IL-1β, while ARG-1 and IL-10 expression were reduced. These data suggest that the NP-pCAR could introduce the anti-CD133 CAR into macrophages and shift their phenotype to the classically proinflammatory M1 subtype.
The effects of this strategy on antitumor responses was tested in the GL261 syngeneic orthotopic mouse glioma model. After tumors were established, these were injected with gene-laden NP (pCAR dose 2 mg/kg). On day 6 after treatment, the CAR-macrophage frequency among all F4/80+ cells was approximately 5%, which increased to about 12% on day 12. In vivo phagocytic cell killing of CD133-expressing tumor cells by NP-pCAR-treated macrophages was observed. Treatment with NP-pCAR or anti-CD47 antibody alone reduced tumor burden, while the combination treatment induced the most tumor regression. The combination treatment group had the highest ratio of healthy brain tissue versus tumor areas. This resulted in an 83% survival rate at day 120, while this combination did not induce severe organ damage or body weight loss.
Long-term survivors in the combination treatment group were rechallenged with glioma cells in the opposite hemisphere to assess whether the treatment had induced immunological memory. The mice remained tumor-free and survived long-term. In these mice, there was an increase in the percentage of effector memory and central memory T cells in the blood and spleen. The mice also had higher levels of serum cytokines, including IFNγ, IL-12, IL-15, and TNFα. These data suggest that the combination treatment induced adaptive immune responses.
To assess the treatment in a situation that would occur in patients after surgery, the researchers made use of a brain tumor resection mouse model, in which 12 days after tumor inoculation with GL261 cells, the visible tumor was surgically removed under a stereoscope. In the tissue surrounding the resection cavity, remaining tumor cells and macrophages were observed. Then, to test the treatment, the cavity was injected with hydrogel formulations containing NP-CARs and/or anti-CD47 antibody. In mice treated with the NP-pCAR gel, there was a decrease in tumor cell bioluminescence signals on day 33 after inoculation. The efficacy was even higher when this treatment was combined with an anti-CD47 antibody, and mice survived until at least day 120. No toxicity was detected in macrophages, neurons, or astrocytes, and there was no sign of gliosis. In the combination treatment group, the tissue surrounding the cavity on day 19 after inoculation contained higher levels of CD8+ T cells and M1 macrophages and lower levels of M2 macrophages than control groups. Furthermore, secretion of IFNγ, IL-12, and TNFα was elevated in the combination group.
To assess the CAR gene delivery efficiency in the human setting, human THP-1 cells and patient-derived glioma (PDG) xenograft models were used. The monocytic THP-1 cells were induced with an M2-like phenotype and treated with the NP-pCAR. More than 31% of cells had detectable EGFP (a fluorescent indicator for CAR transduction). These cells had enhanced phagocytotic activity against tumor cells and upregulated secretion of TNFα and IL-1β, and there was an increase in the frequency of CD80+ M1 macrophages. Finally, an orthotopic GBM model was established, in which PDG cells were intracranially injected into human hematopoietic stem cells NOD.Cg-Prkdcscid Il2rgtm1SugTg(SV40/HTLV-IL3,CSF2)10-7Jic/Jic (HuHSC-NOG-EXL) mice. After the surgical debulking of established tumors, the hydrogel-supported combination treatment was injected into the cranial cavity. This induced tumor growth inhibition, and 83% of mice survived more than 90 days, suggesting this method is also effective in humanized models.
The data presented here reveal a new approach to treating GBM to reduce tumor recurrence after debulking surgery. If these data can be repeated in patients, the retargeting of macrophages and the local induction of adaptive immune responses might improve patient outcomes.
Write up by Maartje Wouters, image by Lauren Hitchings.



