Most tumors exhibit a high degree of genetic and functional heterogeneity, but how that heterogeneity is established and evolves in situ under immune pressure has been difficult to study. To tackle this obstacle, Milo et al. established a mouse model of MYC-driven B cell lymphoma with a large degree of genetic diversity, both between and within individual mice. In this model, chromosomes are frequently lost, which contributes to genetic diversity and subsequent mosaic expression of tumor antigens. In order to study tumor heterogeneity in this mouse model, the researchers developed a multicolor barcoding strategy that allows for analysis and visualization of marked tumor cells from clonally diverse tumors.
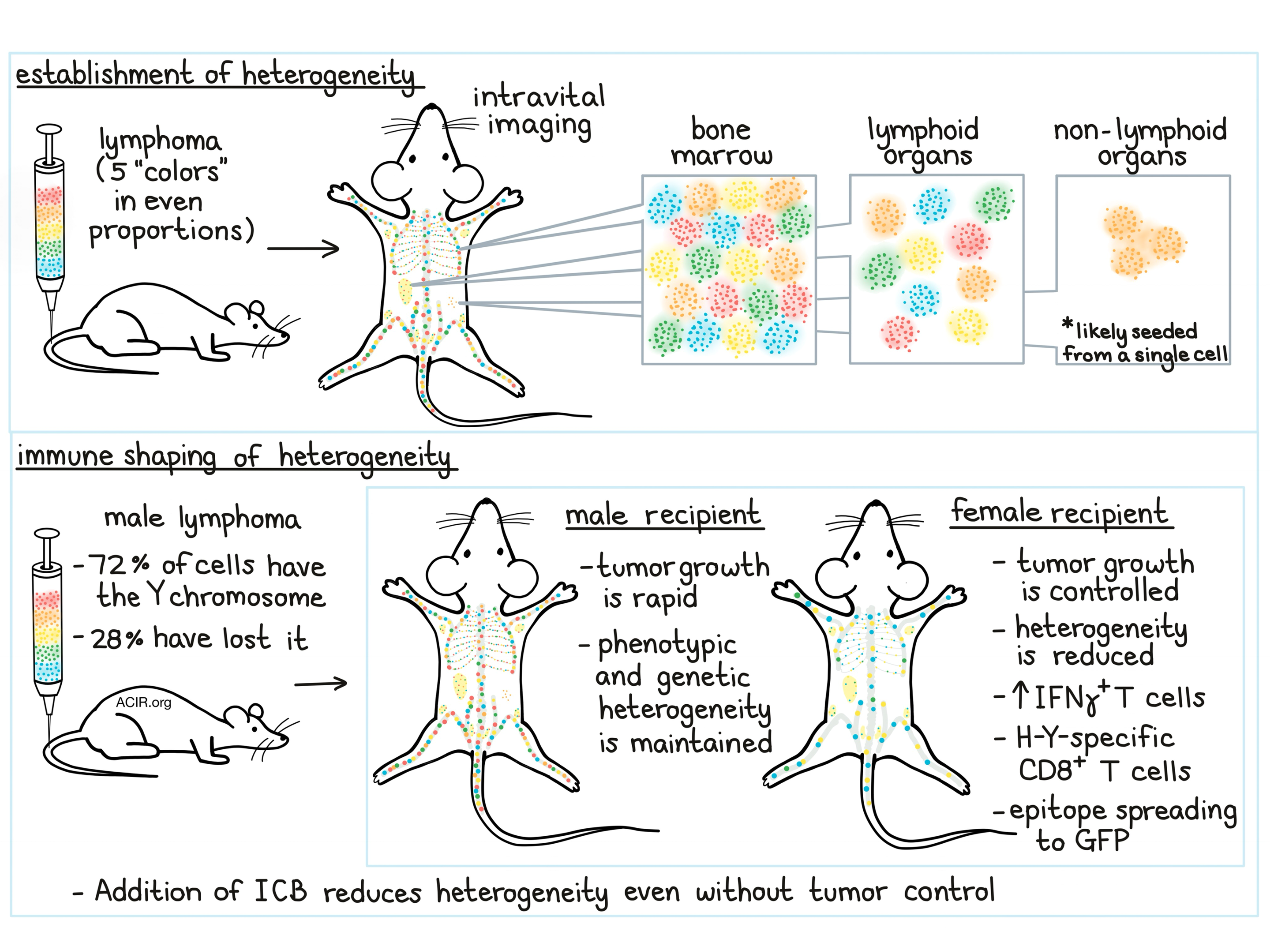
To create their multicolor barcode for lymphoma, Milo et al. used fluorescent proteins (alone or in combination) to tag tumor cell lines from individual tumor-bearing mice. They then created a mixture with equal proportions of five “colors” to be transferred into mice. Using flow cytometry, the researchers tracked the proportions of each color on an “evenness” index to determine whether the proportions remained even, showing maintenance of heterogeneity, or skewed, showing loss of heterogeneity. This multiciolor system also lends itself to intravital imaging, which allowed researchers to visualize spatial organization of individual progeny at different stages of tumor development.
In mice, B cell lymphoma growth was observed primarily in the bone marrow, to a lesser extent in the spleen and some lymph nodes, and occasionally in non-lymphoid tissue. Using the multicolor barcoding system to study the extent of tumor heterogeneity, the researchers found that in most lymphoid locations, all 5 colors could be recovered with comparable “evenness”. Tumor nodules that developed in non-lymphoid tissues, however, were usually only one color. This effect was found to be independent of immune editing (as it also occurred in Rag-/- mice), suggesting that masses in non-lymphoid tissues were seeded from a single cell. Looking closer at heterogeneity just within bone marrow, the researchers found that early on, small patches of cells of each color were established in distinct locations, forming individual niches containing progeny from a single cell. These patches grew over time but showed little migration or overlap.
To better understand how the immune system responds to and shapes intratumor heterogeneity, the researchers utilized a male mouse tumor model in which 28% of tumor cells had lost the Y chromosome. This male lymphoma line was then transferred into either male or female mice, with the expectation that in females, cells expressing the Y chromosome would be highly antigenic. As expected, tumors grew rapidly in males, but were well-controlled for a period of time in females. Using the multicolor barcoding strategy, the researchers found that male mice maintained tumor heterogeneity over time, while females showed a dramatic loss of heterogeneity and the emergence of a few dominant clones. This effect was not observed in Rag2-/- mice, suggesting that it was likely caused by immunoediting. As further evidence of this, the researchers observed an increase in IFNγ-producing T cells in females and identified CD8+ T cells specific to H-Y – a Y chromosome antigen. Further analysis of cell karyotypes showed that tumor cell clones that had maintained the Y chromosome were efficiently eliminated by CD8+ T cells in female mice.
When the researchers restricted karyotype analysis to only the subset of tumor cells that had lost the Y chromosome, they still saw a reduction in diversity in females compared to males. Further, they found that female mice mounted a T cell response to green fluorescent protein (GFP, typically poorly immunogenic), suggesting that epitope spreading – which occurs when the response to strongly immunogenic tumor antigens (such as H-Y on the Y chromosome) enhances responses to poorly immunogenic tumor antigens – may have contributed to the loss of tumor heterogeneity, even among tumor cells lacking a strong immunogenic target.
In order to better quantify genetic changes in intratumor heterogeneity, Milo et al. utilized whole-exome sequencing to identify tumor mutations and predict neoantigens and to track possible changes in these targets under immune pressure. Using the distribution of allele frequencies among targets to measure and calculate heterogeneity, the researchers found that genetic diversity was high in tumors isolated from bone marrow and low in tumors isolated from non-lymphoid tissues, confirming the multicolor barcoding results. They also found that tumors that had been growing under high immune pressure in female mice were less genetically diverse than tumors that had been growing under low immune pressure in males.
Having shown that immune pressure restricts intratumor heterogeneity, Milo et al. investigated the effect of checkpoint blockade on heterogeneity. Treatment with anti-PD-1 in male mice inoculated with male lymphoma had little effect on tumor growth, but significantly reduced tumor diversity and favored clonal dominance. Anti-CTLA-4 alone had little effect, but combination anti-PD-1/anti-CTLA-4 increased mouse survival and significantly decreased tumor heterogeneity, likely through broadening of the immune response. These results were confirmed in a second B cell tumor model. Together, this suggests that checkpoint blockade can impact diversity and tumor composition even in the absence of strong tumor control.
Overall, Milo et al. show that multicolor tumor barcoding can be used to study genetic intratumor heterogeneity and that heterogeneity can be shaped and restricted by the immune response. This strategy also represents a powerful new tool to further study tumor heterogeneity in a broad range of cancers and contexts.
by Lauren Hitchings



