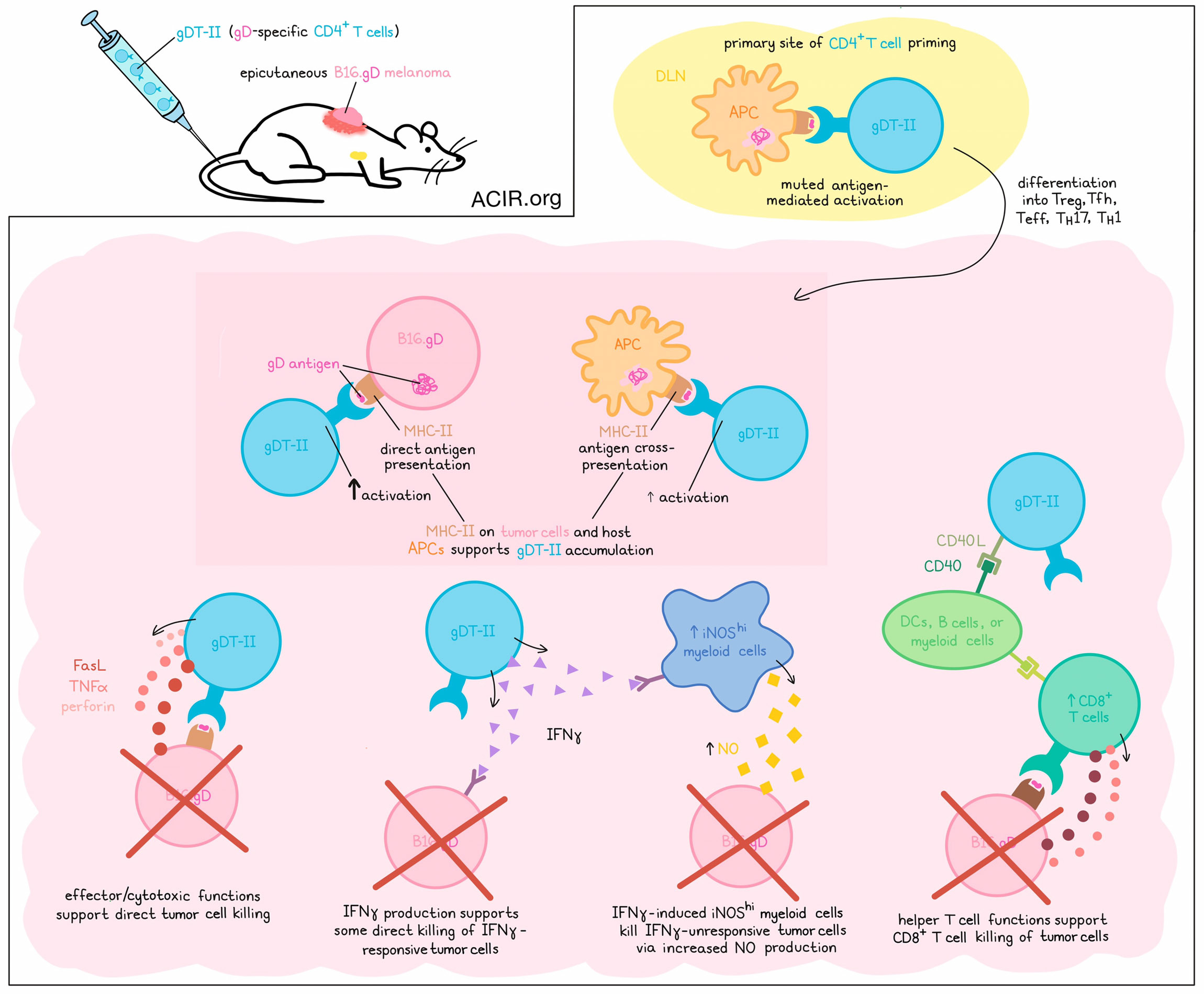
CD4+ T cells are best known for their roles in helping CD8+ T cells, but in recent years, researchers have identified CD4+ T cells across broad ranges of subsets playing roles in activation, recruitment, reprogramming, and direct cytotoxicicity. To better understand these CD4+ T cell roles and their contributions to antitumor immunity, Bawden et al. used an epicutaneous model of melanoma to investigate CD4+ T cell interactions with cancer cells and host APCs, and better understand the roles they play in different contexts. The results were recently published in Science Immunology.
To begin, Bawden et al. utilized B16.F10 melanoma cells engineered to express the glycoprotein D (gD) antigen from herpes simplex virus (HSV). When pre-treated with IFNγ to induce MHC-II expression, these tumor cells could be recognized by gD-targeted TCR-transgenic CD4+ T cells (gDT-II), as could DCs pulsed with B16.gD lysates. Next, the researchers established an epicutaneous model for melanoma, in which B16.gD cells were transplanted into skin abrasions, establishing delayed-onset progressing tumors in around 60% of mice, the majority of which also metastasized to tumor-draining brachial lymph nodes. In mice previously infected with HSV-1 or that received gDT-II before tumor challenge, antitumor immunity was observed.
Evaluating the contributions of CD4+ T cells, the researchers used anti-CD8ɑ antibodies to deplete mice of CD8+ T cells. This partially reduced the protection afforded by gDT-II transfer. Further, transfer of gDT-II Fut7-/- cells with reduced migratory functions also showed limited efficacy against primary tumors, suggesting that infiltration of tumors was important to CD4+ T cell-mediated antitumor immunity. gDT-II were also less effective in mice lacking CD40, suggesting the importance of cross-talk between CD4+ T cells and DCs, B cells, or myeloid cells. These results pointed towards CD4+ T cells playing helper roles in tumors.
Next, the researchers set up a system for tagging and tracking both tumor and gDT-II cells through flow cytometry and intravital imaging. This revealed that brachial lymph nodes were the tumor-draining LNs and primary sites of CD4+ T cell priming early after the inoculation of tumor cells. In B16.gD tumor-bearing mice, gDT-II cell division occurred more slowly, and activation was more limited than in mice exposed to HSV1, suggesting muted antigen-mediated activation in the tumor setting. Further, gDT-II cells in the tdLNs remained higher in tumor-bearing vs. tumor-free mice, suggesting ongoing responses to antigens.
To study gDT-II differentiation, Bawden et al. used scRNAseq and paired TCRseq to evaluate CD44hiCD4+ T cells from lymph nodes, skin inoculation sites, and tumors at 8 and 20 days post-inoculation. In their analysis, 13 clusters were distinct between LNs and the skin/tumor sites. Expanded TCR clones were identified across various CD4+ T cell subsets, with individual TCR clonal families biased for particular differentiation states. Transcriptional analysis revealed evidence of recent activation. Ultimately, the researchers were able to identify clusters of Tregs, Tfh cells, and CD4+ effector cells, including Teff, TH17, and peripheral TH1 clusters. A minor proportion of effector cells also displayed evidence of cytotoxicity, expressing Gzma, Gzmb, Gzmk, and/or Slamf7, but rarely Prf1.
Investigating whether effector CD4+ T cells could directly kill tumor cells, the researchers cocultured B16.gD cells with gDT-II and observed efficient dose-dependent killing of MHC-II-expressing B16.gD. Killing was mostly antigen-specific, and did not require the presence of TRAIL or IFNγ during killing. While perforin and TNFα were found to play minor, non-redundant roles, tumor cell killing was mostly dependent on FasL. When FasL, perforin, and TNFα were inhibited simultaneously, tumor cell killing was almost completely inhibited.
Next looking at MHC-II expression by B16.gD in vivo, the researchers noted that it was restricted to small tumors, which had higher densities of infiltrating CD4+ T cells, and that MHC-II knockdown reduced CD4+ T cell infiltration. In MHC-II-deficient hosts, MHC-II expression by B16.gD was sufficient to activate gDT-II-mediated immunity and support for increased CD8+ T cells. Meanwhile in wild-type hosts, gDT-II were unable to control MHC-II-deficient B16.gD, suggesting that direct priming was more important than cross-priming in activating CD4+ T cell-mediated antitumor immunity. Intravital imaging also showed that some gDT-II moved slowly or stopped when interacting with melanoma cells, providing further evidence for direct antigen recognition.
While MHC-II expression by host APCs alone was not sufficient to control tumors in vivo, in vitro-generated DCs loaded with antigens could induce gDT-II killing of MHC-II-deficient B16.gD in vitro, suggesting that APCs could contribute to activation, and that MHC-II expression on melanomas was not required for them to be killed. Similar results were observed in vivo, where tumor debris-laden CD11c+ APC accumulated around tumors, ingested debris, expressed costimulatory molecules, and directly interacted with gDT-II, suggesting at least some APC-mediated activation of CD4+ T cells. Similar patterns were identified in samples of human melanomas.
In an effort to understand the effector pathways involved in CD4+ T cell-mediated antitumor immunity, the researchers used mice lacking endogenous lymphocytes and chose an intermediate dose of gDT-II that protected about 90% of mice from tumor development. In this model, gDT-II also protected against MHC-II-deficient tumors, suggesting host APC-mediated protection. Next, the researchers interfered with IFNγ-, TNFɑ-, FasL-, and perforin-dependent pathways using genetic deletions or blocking Abs. While deleting perforin had little effect, blocking either TNFɑ or FasL slightly reduced antitumor efficacy – an effect that was enhanced when TNFɑ and FasL were both blocked. Finally, blocking IFNγ almost completely abolished gDT-II-mediated protection, causing early tumor outgrowth in nearly all mice. Similar results were observed in IFNγ-deficient gDT-II, confirming gDT-II as the source of the protective IFNγ. Further, the protective effect was only slightly dependent on the melanoma cells expressing IFNγR, suggesting yet another mechanism was at play.
Following recent evidence that IFNγ can induce iNOS expression and NO production in myeloid cells, the researchers found that transfer of WT gDT-II induced iNOShiMHC-II+Ly6C+Ly6G− monocytic cells in the majority of mice. Transfer of IFNγ-deficient gDT-II induced 10-fold lower frequencies of iNOShi myeloid cells. In mice with wild-type B16.gD, the iNOS inhibitor L-NIL did not affect gDT-II protection, but in mice with B16.gDIfngr1KO tumors, L-NIL allowed for tumor outgrowth in six of eight mice despite transfer of gDT-II, suggesting that myeloid-derived NO may play a role in CD4+ T cell-mediated suppression of IFNγ-unresponsive melanomas.
Overall, these results reveal a wide range of functions of antitumor CD4+ T cells, which can be activated by MHC-II-expressing host APCs and/or melanoma cells. Depending on different contexts, CD4+ T cells can perform helper functions, direct killing of tumor cells via TNFα- and FasL -mediated cytotoxicity, and production of IFNγ, which acts both directly on melanoma cells and indirectly by inducing nitric oxide synthase in myeloid cells.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Emma Bawden answered our questions.

What was the most surprising finding of this study for you?
With an initial focus on CD8+ T cells, we set out to study CD4+ T cell “help” in our recently developed, orthotopic melanoma model. We did not anticipate that CD4+ T cells would have such a potent antitumoral role, independent of regulating CD8+ T cells. We were initially surprised that the transfer of a very low dose of naive, antigen-specific CD4+ T cells (which was selected to mimic an endogenous CD4+ T cell response) prevented mice from developing tumors. In addition, we were surprised by the number of redundant mechanisms employed by CD4+ T cells to control melanoma. In fact, the redundancy of these pathways rendered them difficult to elucidate because blocking one effector pathway in vivo (by gene deletion or blocking antibody) did not always present an obvious phenotype. The loss of CD4+ T cell-driven protection in some instances required blocking several effector pathways simultaneously. Finally, contrary to what has been suggested in the literature, we found that the perforin-dependent killing pathway, a dominant effector mechanism of CD8+ T cells, was not employed by CD4+ T cells to control melanoma in our model.
What is the outlook?
Now that we have discovered mechanisms by which CD4+ T cells target tumors, we would like to further investigate the pathways that regulate CD4+ T cell differentiation and function. This is highly clinically relevant because CD4+ T cells can differentiate into many different subsets with different functions, including T regulatory cells (Treg) that suppress antitumor immunity. Knowing how to shift the balance from Treg to antitumor T effector cells is critical for a productive immune response against tumors. Our goal is to unravel the complex biology of CD4+ T cells so that these cells can be harnessed in cancer immunotherapies.
What was the coolest thing you’ve learned (about) recently outside of work?
I’m learning about music video production and I recently acted as an FBI agent in the video clip of my favorite band, Fan Girl: https://www.youtube.com/watch?v=1ejlsBjHmAw



