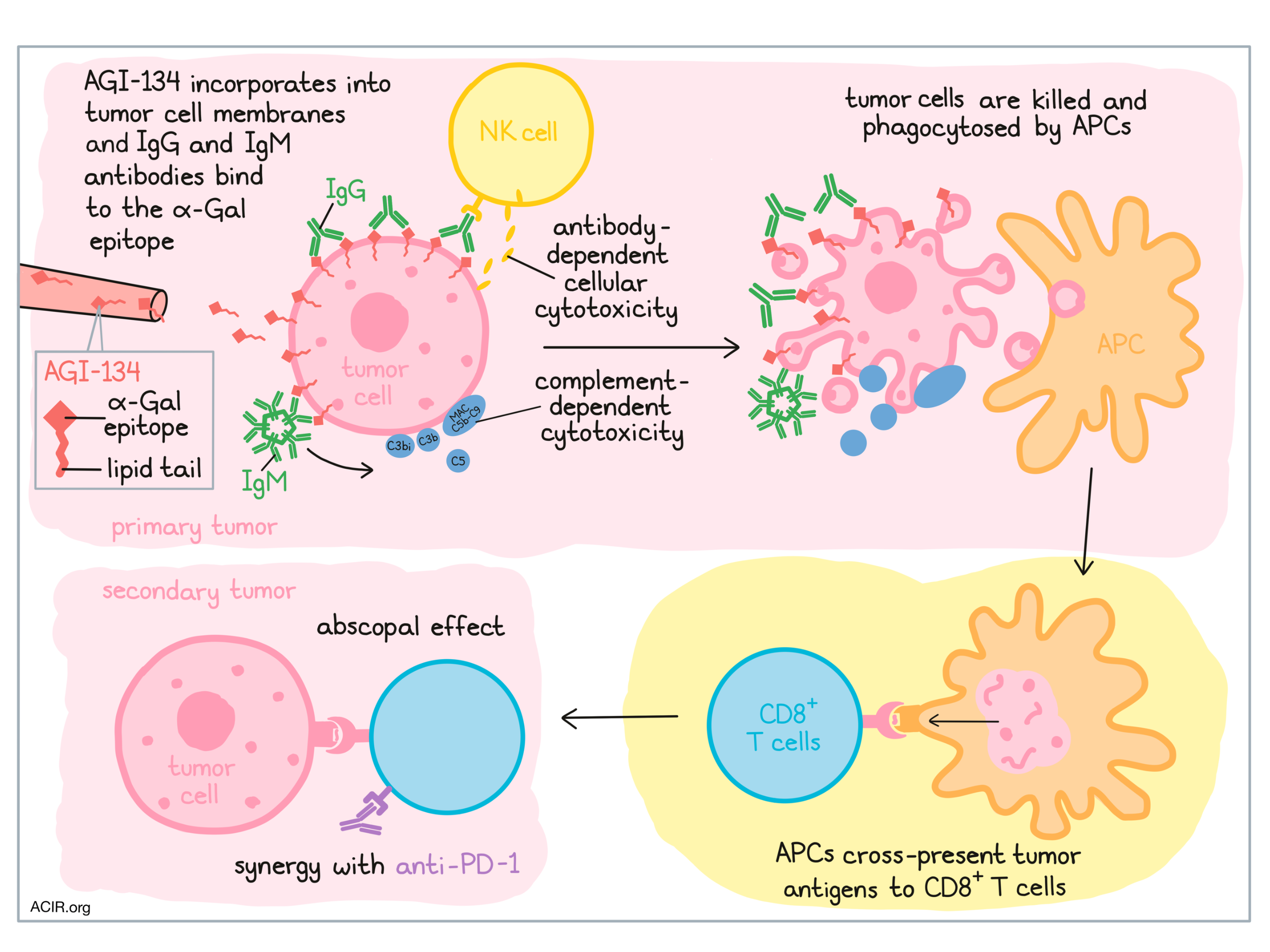
The cancer immunity cycle starts and ends with cancer cell death, and inducing cancer cell death at the site of a tumor to kickstart antitumor immunity is a developing strategy in cancer immunotherapy. One approach that has been explored to induce cancer cell death in situ is the introduction of α-Gal epitopes, which are readily recognized by naturally occurring human antibodies. Previous studies have found that an α-Gal glycolipid preparation extracted from rabbit erythrocytes effectively killed cancer cells and kickstarted the cancer immunity cycle, however, developing a human therapy from a crude biological extract is not particularly practical. To overcome this problem, Shaw et al. tested the functionality of the fully synthetic, easy-to-manufacture, α-Gal glycolipid-like molecule, AGI-134; the study results were recently published in Cancer Cell International.
To establish the functionality of AGI-134, which is comprised of a lipid tail linked to the α-Gal epitope, Shaw et al. began with a series of in vitro experiments. First, human SW480 and A549 adenocarcinoma cell lines were treated with AGI-134. Antibodies to α-Gal are naturally abundant in humans due to antibody responses to similar molecules in the gut microbiome, and treatment with AGI-134 was shown to enhance the binding of anti-Gal IgG and IgM antibodies to the surface of tumor cells, suggesting that AGI-134 incorporates into the cancer cell membranes and presents α-Gal on the cell surface.
Given that IgM antibodies are potent activators of the classical complement pathway and that IgG antibodies can activate antibody-dependent cellular cytotoxicity (ADCC) by NK cells, Shaw et al. investigated both of these pathways. A549 and SW480 cells treated with AGI-134 were killed when incubated in normal human serum (NHS), and the killing of SW480 cells was shown to be complement-dependent. In the A549 cells, the researchers observed that AGI-134 induced deposition of complement C3b/C3bi and formation of membrane attack complex (MAC) C5b-C9 on A549 cells, and increased the concentration of C5 (a powerful immune stimulant) in the supernatant, indicating complement activation. ADCC was evaluated using a reporter assay and a human NK cell assay, which revealed an increase in FcγRIIIa activation and induction of NK cell-mediated ADCC following treatment with AGI-134.
To determine whether the cell death mediated by AGI-134 and NHS could induce an antitumor immune response, Shaw et al. explored whether cells and cellular debris (complexed with anti-Gal and complement) were internalized and processed by antigen-presenting cells (APCs). Using fluorescent tags, they showed that human monocyte-derived macrophages phagocytosed AGI-134-treated A549 cells at a higher rate than untreated A549 cells. Similarly, murine CD8α+ DCs, which express MHC-I and cross-present antigens to CD8+ T cells, were also capable of phagocytosing AGI-134- and NHS-treated target CHO-K1 cells.
To show that antigens from the phagocytosed target cells could be cross-presented to CD8+ T cells, the researchers generated CHO-K1 cells expressing OVA linked to a fluorescent tag. Administration of AGI-134 and NHS to these cells induced cell death, and the dead cells were incubated with CD8α+ DCs in different ratios. After a period of incubation, OT-I T cells (recognizing OVA) were added to the mix, and their activation was measured by IFNγ production. Increased OT-I activation correlated with the increased ratio of dead cells to DCs, indicating cross-presentation of tumor-associated antigens. Of note, OT-I T cell activation required DC expression of DNGR-1, the molecule on CD8α+ DCs responsible for the uptake of dead cells and initiation of cross-presentation.
Overall, in vitro testing showed that AGI-134 incorporates into cancer cell membranes, leading to presentation of the α-Gal epitope on the surface of the cancer cells. In the presence of NHS, this epitope is recognized by IgG and IgM antibodies, inducing ADCC and activation of the complement system, both of which induce cell death. Killed cells can then be phagocytosed by APCs, which may cross-present tumor-associated antigens and activate antigen-specific CD8+ T cells. Based on the in vitro evidence for this mechanism, Shaw et al. hypothesized that AGI-134 could be used to kickstart the cancer immunity cycle in vivo.
To test the antitumor efficacy of AGI-134, the researchers used α1,3GT-/- mice, which, like humans, do not express α-Gal epitopes; the mice were immunized to induce production of anti-Gal antibodies. For tumor lines, B16F10 and JB/RH melanomas were chosen, as they too lack expression of α-Gal. To ensure that this model would be effective, the researchers confirmed that AGI-134 incorporated into the cell membranes of B16F10 and JB/RH cells, enhanced the binding of mouse-derived IgG and IgM antibodies to the cancer cells, and induced the deposition of complement proteins (C3b and MAC).
In α1,3GT-/- mice bearing B16F10 melanoma tumors, the researchers showed that intratumoral (i.t.) injection of a glycolipid (representative of AGI-134) distributes throughout the tumor, and that i.t. injection of AGI-134 induced complement activation within the tumor. Two intratumoral doses of AGI-134 delivered 24 hours apart induced significant tumor regression compared to control. In a dual-flanked mouse model, a single injection of the primary lesions also impaired the development of the uninjected tumor, with contralateral tumors developing in 86% of PBS-treated mice, but in only 16% of AGI-134-treated mice. This abscopal effect was found to be dependent on expression of anti-Gal antibodies. Overall, the antitumor efficacy of AGI-134 was found to be dose-dependent, and long-term experiments showed that a single dose could protect against contralateral tumor development for over 90 days of observation. Similar antitumor effects were observed in the JB/RH tumor model; in this model, AGI-134 also gave mice a survival benefit.
Following the hypothesis that the observed abscopal effect was due to activation of tumor antigen-specific CD8+ T cells, Shaw et al. investigated whether AGI-134 might synergize with PD-1 blockade. Again using B16F10 tumors in α1,3GT-/- mice bearing B16F10 tumors, the researchers identified suboptimal doses of each monotherapy (AGI-134 and anti-PD-1). When these suboptimal doses were used in combination, only 6% of mice developed a distal tumor within the 35 day observation window, compared to 38% of mice treated with suboptimal dose AGI-134, 62% of mice treated with a suboptimal dose of anti-PD-1, and 77% of mock-treated mice. Based on these results, a phase I/IIa clinical trial of AGI-134 and AGI-134 plus anti-PD-1 has been initiated for patients with unresectable metastatic solid tumors.
by Lauren Hitchings
MEET THE RESEARCHER
This week, Sascha Kristian and Arik Zur answered our questions.

What prompted you to tackle this research question?
While the use of checkpoint inhibitors (CPIs), antibodies that unleash the antitumoral activity of pre-existing T cells, has revolutionized cancer therapy, patients with tumors that have little inflammation or T cell infiltration are unfortunately mostly refractory to CPI treatments. Agalimmune, which was acquired by BioLineRx in 2017, thought to develop a fully synthetic drug that could enhance intratumoral inflammation, improve patient-specific, tumor-specific antigen processing and T cell generation. If successful, such drug could serve as a novel immunotherapy that could be used alone or in combination with CPIs, especially in patients who are otherwise refractory to CPI treatment. Our proof-of-concept study with AGI-134 suggests that intratumoral delivery of α-Gal glycolipids to treat solid tumors is a promising approach to induce potent patient-specific antitumor immune responses. AGI-134 harnesses a pre-existing immune machinery using the α-Gal antigen, and may serve to universally convert solid tumors to personalized anti-cancer vaccines. We envisioned this study to be an enabling first step on the way to testing the clinical utility of α-Gal glycolipids in patients with solid tumors and it superseded our expectations. BioLineRx is currently in the midst of a Phase I/IIa clinical study testing intratumoral injections of AGI-134 in patients.
What was the most surprising finding of this study for you?
When we started our proof-of-concept studies, we were quite surprised to see how well a single intratumoral injection of AGI-134 worked in the quite aggressive mouse B16F10 melanoma model. Specifically, AGI-134 had a strong abscopal effect, i.e., the intratumoral treatment of a primary tumor with AGI-134 potently protected mice from the growth of secondary, uninjected melanoma lesions. We were very encouraged to see that the effect was reproducible and was also observed in a second melanoma model with JB/RH cells. In addition, AGI-134 and an anti-PD-1 immune checkpoint inhibitor have shown to work synergistically, suggesting that AGI-134 may be an excellent combination partner for CPIs in cancer patients, offering the potential to broaden the utility of such immunotherapies and improve the rate and duration of responses in multiple cancer types.
What was the coolest thing you’ve learned (about) recently. outside of work?
AZ: I have recently become a parent to a smiley and chatty baby girl. All of my free time outside of work is dedicated to the little creature, attending to her needs. As a result, the main thing I learned in the last few months is that I can survive and function with significantly fewer sleeping hours each day!



