Both PD-1 and TIGIT are checkpoint molecules known to inhibit the antitumor effects of CD8+ T cells, and blocking them in combination has been shown to have synergistic immunotherapeutic effects. However, the exact mechanism and dynamics of this synergy are incompletely understood. In recent research, Nutsch, Banta, and Wu et al. utilized a multicompartment, multi-omics, single-cell approach to explore the individual and combined effects of anti-PD-L1 and anti-TIGIT on T cells in the draining lymph nodes (dLNs), tumors, and blood of mice. Their results were recently published in Nature Cancer.
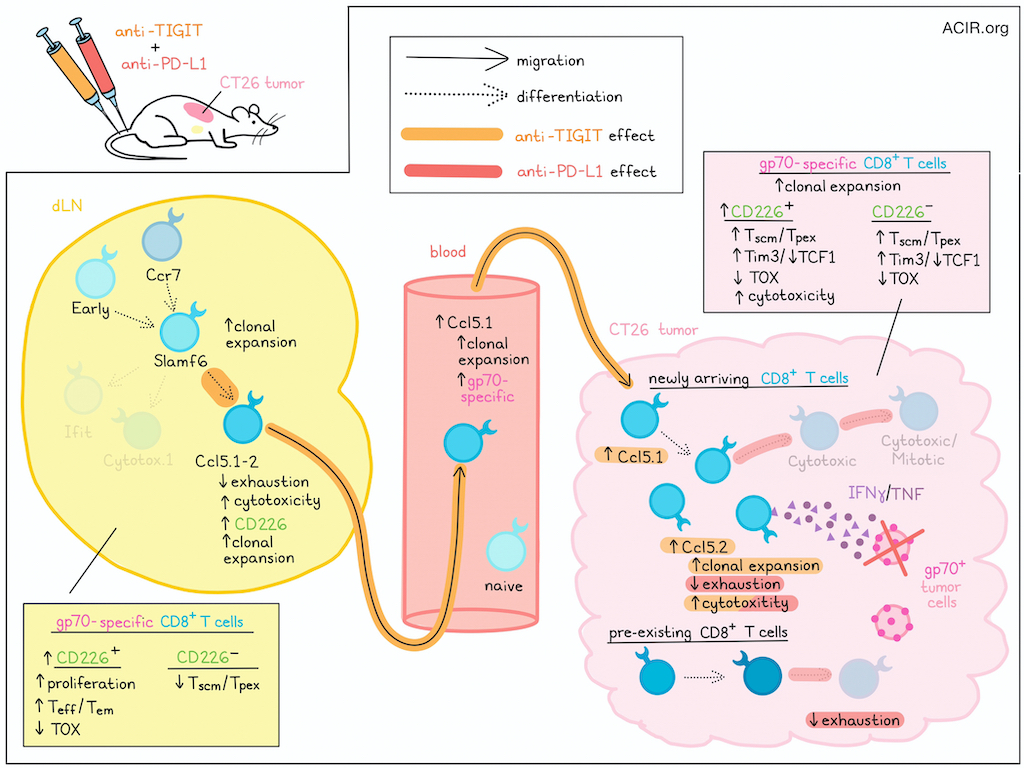
To study the effects of anti-PD-L1 and anti-TIGIT, the researchers utilized a CT26 syngeneic mouse tumor model, which could be effectively controlled with combination therapy, with increased tumor antigen (gp70)-specific CD8+ T cells in dLNs and blood. Blocking T cell egress from lymph nodes using FTY720 during combination therapy increased the accumulation of gp70-specific CD8+ T cells in dLNs, eliminated gp70-specific CD8+ T cells from the blood, and reduced antitumor immunity (despite local expansion gp70-specific TILs), suggesting that migration of antigen-specific CD8+ T cells from the dLN was critical to antitumor immunity.
Next, looking at the role of CD226, a stimulatory counter-receptor to TIGIT, which also binds the TIGIT ligand PVR, and which has previously been found to be intracellularly suppressed by PD-1, the researchers found that anti-TIGIT alone or in combination with anti-PD-L1 increased the frequency of CD226+gp70+CD8+ T cells in both dLNs and tumors, regardless of FTY720 treatment. Delving into the phenotypes of these cells, the team saw that only combination treatment increased the proliferation and Teff/Tem phenotypes in CD226+gp70+CD8+ T cells while reducing the frequency of Tscm/Tpex cells among CD226-gp70+CD8+ T cells in dLNs. In tumors, however, anti-TIGIT or combination treatments increased Tscm/Tpex cells within both CD226- and CD226+ gp70+CD8+ T cell populations, along with more differentiated iterations of these subsets, which expressed Tim3 and lost expression of TCF1. Similarly, combination treatment decreased in the expression of TOX in CD226+ populations in dLNs and in both CD226- and CD226+ populations in tumors. However, increased cytotoxicity (IFNγ and TNF) was only observed among CD226+ populations in tumors, and this effect was lost upon treatment with FTY720, suggesting that T cells derived from the periphery might have stronger effector functions in tumors. Further, blockade of CD226 reduced the frequency of dLN-derived antigen-specific subsets, abrogated the reduction of TOX expression, and impaired the antitumor efficacy of anti-PD-L1 and anti-TIGIT combination treatment, confirming that efficacy was dependent on CD226-mediated costimulation.
Using scRNAseq, scTCRseq, CITEseq, and ADTseq (Antibody-Derived Tag sequencing; labels T cell receptors using tetramers for gp70, as well as other non-gp70 antigens) on T cells from tumors, dLNs, and blood of experimental mice, Nutsch, Banta, and Wu et al. identified 24 distinct clusters among 245,675 total T cells and then 20 clusters among 155,496 T cells with high CD8 expression, which had accounted for most of the antigen-specific effector T cells. Two clusters, identified as Ccl5.1 and Ccl5.2, expressed CCL5 and resembled cytotoxic and proliferating cytotoxic clusters, but with reduced signs of exhaustion, resembling previously described “better effectors", stem-like clusters, or transitory exhausted clusters. Further, CD226 was highly characteristic of the Ccl5.1 subset, which was also the only major non-naive cell state identified in the blood.
Next, the researchers used RNA velocity-based trajectory analysis to evaluate differentiation patterns. In dLNs across treatment groups, Early and Ccr7 clusters differentiated into Slamf6 cells, which then differentiated into Ifit or Ccl5 cells. In tumors, Ccl5 cells differentiated into Cytotoxic and then Cytotoxic/Mitotic cells, while another differentiation pathway generated mitotic cells. Evaluating clonal T cell expansion, the researchers found that in the dLNs, most clones were singletons, but clonal expansion was induced in the Slamf6, Ifit.3, and Ccl5.1 clusters following combination treatment. In tumors, most clones (both gp70-specific and non-specific) were expanded, with preferential expansion in Ccl5.2 cells and gp70-specific cells upon combination treatment. In the blood, anti-TIGIT or combination treatments induced the emergence of expanded antigen-specific T cells, which fell into the Ccl5.1 cluster. Using FTY720, the researchers showed that Ccl5.1 cells in the blood likely originated from Slamf6 cells in dLNs, and seeded gp70-specific T cells in tumors, which expanded and differentiated alongside pre-existing gp70-specific and non-specific TILs.
Looking more closely at T cell clonotypic expansion across different tissue sites and phenotypes, the researchers found that anti-TIGIT, anti-PD-L1, and combination therapies each drove distinct differentiation pathways in dLNs and tumors. Overall, anti-TIGIT appeared to support differentiation of dLN Slamf6 cells towards Ccl5.1 cells (rather than towards Cytotox.1 cells) and then promote the migration of Ccl5.1 cells to blood and tumors. Meanwhile, anti-PD-L1 mainly shifted differentiation patterns of T cells within tumors, including in anti-TIGIT-derived Ccl5.1 cells, driving differentiation towards more cytotoxic and less terminally exhausted phenotypes.
To translate these findings from mice to patients, Nutsch, Banta, and Wu et al. analyzed scRNAseq data of peripheral blood T cells from a phase 1b study of anti-TIGIT (tiragolumab) plus anti-PD-L1 (atezolizumab) (T + A) in patients with NSCLC. Mapping this data onto the nearest mouse reference clusters, the researchers found that compared to non-responders (SD/PD), responders (CR/PR) had increased CD8+ T cells mapping to Ccl5.1 and Ccl5.2 clusters, and decreased cells mapping to Ccr7.3 and Ccr7.4 clusters. In bulk scRNAseq data of baseline tumors, Ccl5.2 and Cytotox.2 gene signatures were higher in responders. Strong Ccr7.3, Slamf6, and Ccl5.1 gene scores, and low Cytotox.1 and Cyt/Mit.2 scores were associated with improved hazard ratios for overall survival. Grouping patients according to high versus low expression of the Ccl5.2 gene signature also correlated with OS. High expression of CXCR3, CXCR6 and CCL5 were also associated with response and OS. Thus, the results in mouse were largely replicated in the human setting.
Overall, these results suggest that anti-TIGIT drives the expansion of a unique T cell population, which migrates through the blood to tumors, where anti-PD-L1 alters differentiation, together supporting strong antitumor immunity. This study also suggests antigen-specific CD8+ T cells newly arriving from dLNs may be of higher quality than pre-existing antigen-specific CD8+ T cells in tumors, and may better predict clinical responses and survival following combination treatment.
Write-up and image by Lauren Hitchings



