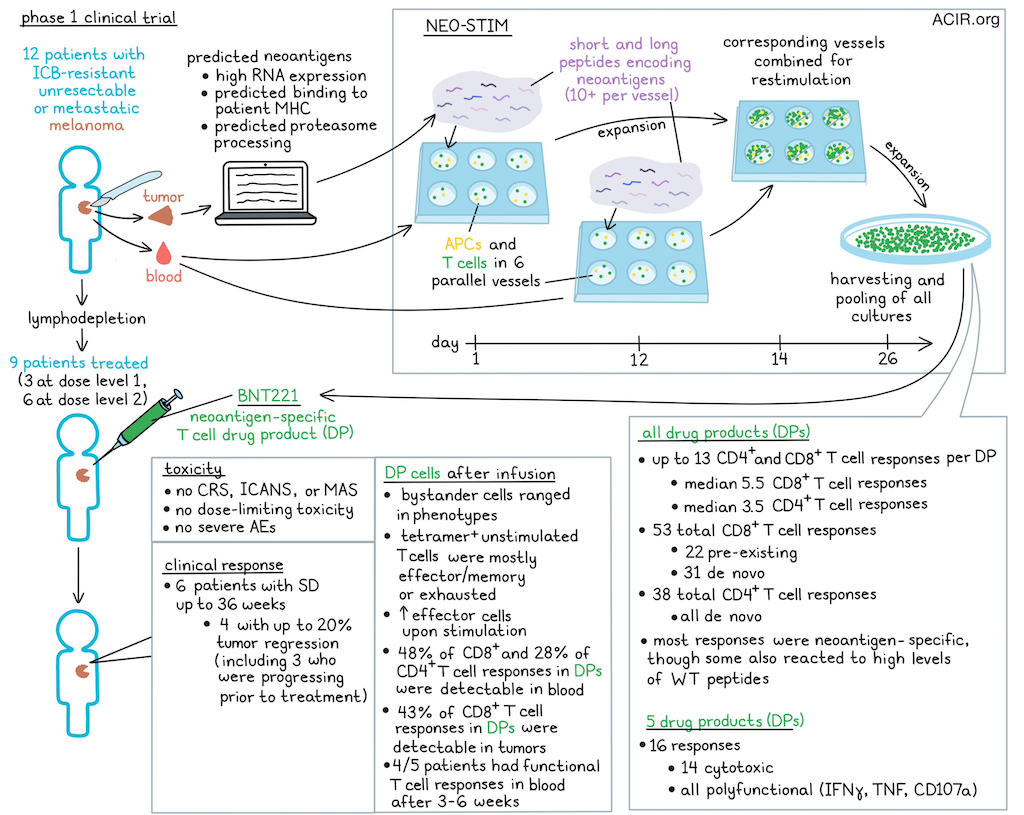
While major progress has been made in targeted and immune checkpoint blockade (ICB) therapy for melanoma, many patients do not respond or their response is not durable. To investigate a new approach for these patients, Borgers et al. performed a Phase 1 study assessing targeted adoptive T cell therapy (ACT) directed against patient-specific neoantigens (BNT221). Their encouraging results were recently published in Nature Medicine.
To identify neoantigens to target, whole-exome sequencing (WES), RNAseq, and algorithms were applied to each patient’s tumor tissue. Neoantigens with high RNA expression, predicted binding to patient-specific MHC, and predicted proteasome processing were selected. An ex vivo induction process, named NEO-STIM, was conducted using short (average 35 per patient) and long (average 13 per patient) peptides encoding the selected neoantigens to prime, activate, and expand neoantigen-specific T cell drug products (DPs). To develop a polyclonal T cell product targeting several epitopes, six parallel culture vessels of patient PBMCs containing autologous antigen-presenting cells (APCs) and autologous peripheral T cells, FLT3L, and (added one day later) ≥10 of the selected peptides were used for priming in each culture vessel. Further, to expand already primed neoantigen-specific T cells, a second PBMC culture was initiated at day 12 and was used for restimulation of the first culture on day 14. On day 26, all cultures were harvested and pooled. Average manufacturing time (needle to needle) was 19.8 weeks.
In this Phase 1 trial, patients with unresectable or metastatic melanoma refractory to ICB were treated with DP infusion after lymphodepleting chemotherapy. DPs were manufactured for 12 patients, of whom three were not dosed due to progressive disease. Three patients received dose level 1 (1x108-1x109 cells) and six patients dose level 2 (2x109-1x1010 cells).
No cases of cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), or macrophage activation syndrome (MAS) were observed. There were no dose-limiting toxicities or related serious adverse events.
As the best overall response, 6 patients had stable disease (SD), of which one lasted up to 36 weeks. Four patients with SD had up to 20% tumor regression, even though 3 of them had significant progression in the period before infusion.
Translational analyses were performed on the DPs and pre- and post-infusion peripheral blood and tumor. First, the researchers assessed the CD8+ and CD4+ T cell responses in the DPs using tetramers for CD8+ T cells, an overnight coculture assay (recall assay) for CD4+ T cells, and neoantigen peptide-loaded APCs. By assessing cytokine production, the researchers found that the NEO-STIM process resulted in several CD8+ and CD4+ T cell responses of various magnitudes in all DPs. Up to 13 T cell responses were generated per DP, with medians of 5.5 CD8+ and 3.5 CD4+ T cell responses.
To investigate whether the generated T cell responses were de novo (not detectable in tumor or blood before treatment) or pre-existing (detectable before treatment), CD8+ neoantigen-specific T cells were isolated from the DP, and single-cell GEX/VDJ/CITE sequencing was performed to phenotype and identify TCR clones. Bulk TCRseq was performed on pre- and post-infusion blood and tumor samples to track these clones. Overall in all DPs, 53 CD8+ responses were detected, of which 22 were pre-existing and 31 were de novo, while all 38 CD4+ responses were de novo. However, the number of pre-existing responses may be underestimated due to low frequencies in the blood pre-treatment.
To assess whether T cells specifically targeted the mutant and not WT epitopes, recall assays were performed. All responses were mutant-reactive over a broad range of EC50s , and 8 showed reactivity to WT peptide, but only at high concentrations. Neoantigen-specific CD8+ T cells from 5 DPs were assessed for cytotoxicity, revealing 14 of 16 tested responses were cytotoxic. All T cell responses were characterized by multiple markers of functionality (production of IFNγ or TNF, or CD107a degranulation). A large subset of the CD8+ responses exerted at least two functionalities (mostly IFNγ and CD107a), and among CD4+ T cells, IFNγ and TNF responses were most common.
There was high overlap in PD-1 expression between various T cell subsets after infusion. Bystander cells (not neoantigen-specific) had a range of phenotypes, including naïve, central memory, effector memory, and effector-like, while tetramer+ unstimulated T cells were mostly effector memory or exhausted-like. Stimulating those tetramer+ cells resulted in increases in effector phenotypes, and preservation of exhausted-like cells. Additionally, there was a wide range of TCR functional avidities (nanomolar to multiple micromolar) among neoantigen-specific TCRs isolated from the DPs.
Next, the researchers addressed persistence, a common determinant of cellular therapy efficacy. Except for one patient, all other patients had measurable DP-specific T cell responses after infusion. Overall, 48% of CD8+ and 28% of CD4+ T cell responses detected in DP were also detected in blood after infusion, and 43% of CD8+ T cell responses were also detected in the tumor. DP-specific T cells in the tumor were from those generally present at the highest frequency in the DP. Functionality of responses at 3-6 weeks after infusion was evaluated, and 4/5 patients had functional T cell responses in the blood. PD-1 was upregulated on most of the tetramer+ CD8+ and CD4+ T cells and these were primarily central or effector memory cells.
Tumor reductions occurred directly after infusion, although there was no clear correlation between any immune marker (dose, fraction of vaccine neoantigen-specific T cells in the DP, number of immunogenic epitopes or T cell functionality) of response. One patient with the greatest tumor reduction was assessed in more detail. Six weeks after infusion, this patient had a 20% reduction of the target lesion and a significant reduction in non-target lesions, including in the lymph node and lungs. Increased T cell infiltration could be observed in the tumor tissue after treatment. The patient had MAGEB278:S>F-specific T cell responses, and six clonotypes were detected in the DP. Of these, four were detected pre-infusion in the tumor, and three were also detected post-infusion. The frequency of these three clonotypes increased 6-fold after infusion in the tumor and ~44-fold in the blood. This was the only patient with multiple responses in whom post-infusion expansion could be detected in both blood and tumor.
The data from this Phase 1 study show that this treatment can induce tumor-specific T cell responses resulting in tumor regression in heavily pretreated patients. Further research will have to establish the characteristics of those responding and how to improve efficacy. Improving T cell functionality of the DP or combining DP treatment with treatment targeting immune inhibiting factors in the tumor microenvironment are interesting strategies to accomplish this goal. A combination with ICB is currently being assessed in the clinic.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Jessica Borgers and lead author Marit van Buuren answered our questions.

What was the most surprising finding of this study for you?
The fact that we were able to generate a highly polyclonal product from PBMCs with multiple CD8+ and CD4+ T cell responses that were both pre-existing and de novo was surprising. We found that these T cells trafficked to both the peripheral blood and the tumor post-infusion, and remained functional.
We saw that the overall manufacturing time posed a challenge for inclusion in the study of these patients with advanced disease, but the fact that we were able to manufacture a drug product for all enrolled patients was a great outcome for this novel adoptive cell therapy.
What is the outlook?
We observed early clinical signals, which was encouraging, but additional development is warranted to simplify the manufacturing process and increase potency, persistence, and proliferative capacity of the generated cell product. Therefore, we are currently testing BNT221 and anti-PD-1 in the clinic, to see whether this combination therapy can enhance clinical efficacy. We are also focusing on methodology to improve the timing and quality of the cell product.
What was the coolest thing you’ve learned (about) recently outside of work?
JB: After my PhD at the Netherlands Cancer Institute (where the patients of this trial were enrolled), I moved to Colorado for a postdoc at the University of Colorado Anschutz Medical Center to get more experience in translational immuno-oncology research. Furthermore, I recently got the opportunity to represent the Netherlands at the Roundnet World Championship in London, which was a really cool experience!
MvB: Recently, my daughter (6 years old) and I have spent a lot of time exploring the woods in the area where we live in Massachusetts, USA. I’ve learned that little things, such as deer tracks in the snow or the thickness of the ice can make for awesome together time that we both enjoy!



