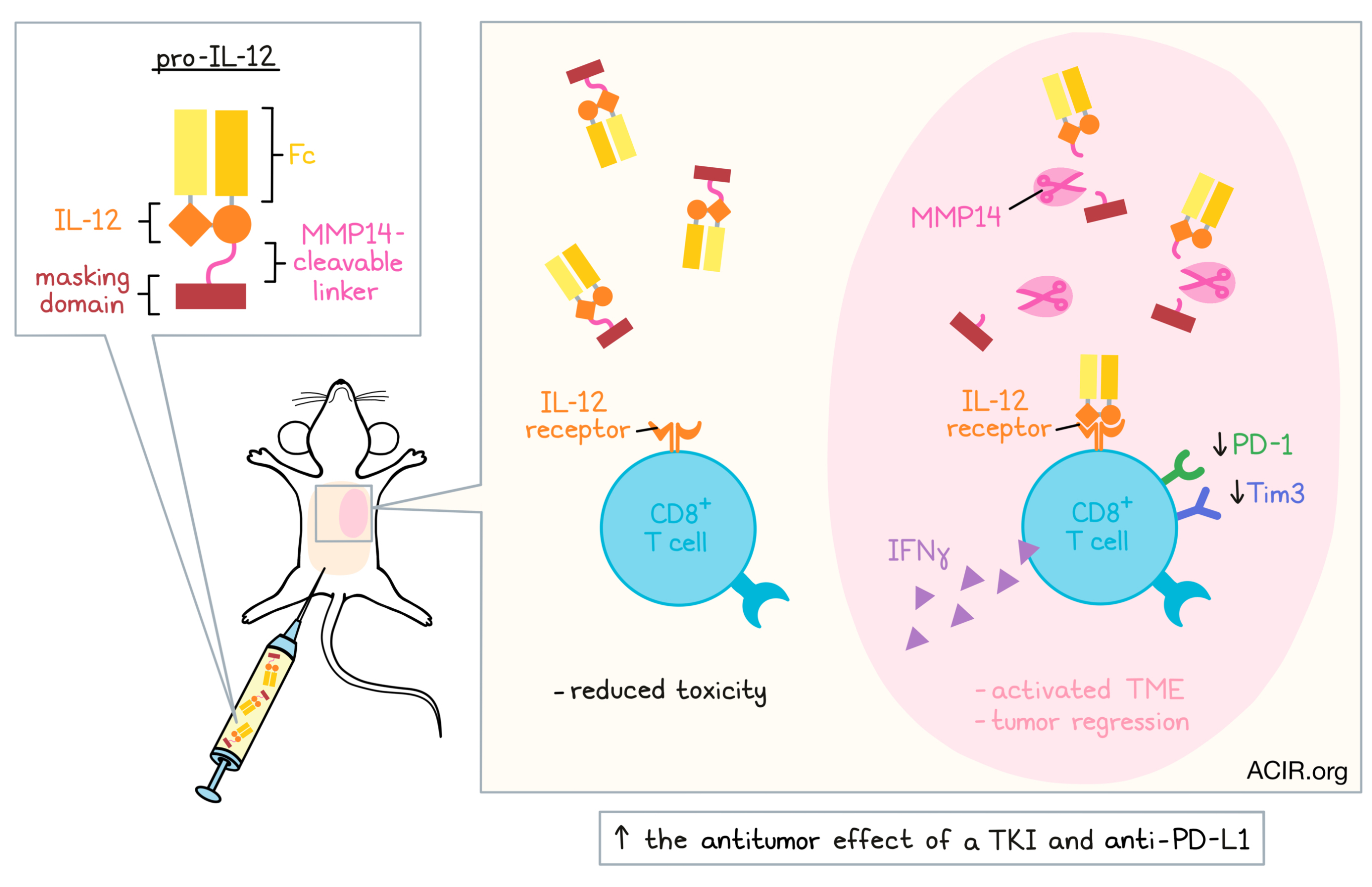
Strategies to activate tumor-infiltrating lymphocytes (TILs) can support existing cancer therapies and overcome resistance mechanisms such as exhaustion. One such approach, the inflammatory cytokine IL-12, can activate T cells and may be linked to patient survival, but its application is limited by toxicity and a short half-life. Recently published in Science Immunology, Xue and Moon et al. overcame these drawbacks by engineering an improved IL-12 therapy.
Xue, Moon, and the team began by increasing the stability of IL-12 in circulation. To do so, they linked IL-12 to a human Fc (IL-12–Fc) in two forms: a heterodimer and homodimer. These modifications did not alter IL-12 bioactivity (via induction of STAT4 signaling in a reporter cell line), and in fact, improved MC38 antitumor efficacy over recombinant mouse IL-12. Hetero-IL-12–Fc more effectively treated MC38 and B16F10 tumors and maintained its serum concentration compared to homo-IL-12–Fc, and was selected for further studies. However, both versions induced inflammatory serum cytokines after i.p. delivery.
To tackle the toxicity issue, the researchers aimed to formulate hetero-IL-12–Fc as a prodrug that would ideally circulate in an inactive state, but become activated in the tumor microenvironment (TME). Matrix metalloproteinases (MMPs), a class of enzymes overexpressed in the TME, presented an opportunity to enable tumor-specific IL-12 activity through their local enzymatic activity. The team linked hetero-IL-12–Fc through an MMP14-cleavable peptide to a masking domain, based upon the IL-12 receptor, which would already bind IL-12 with high affinity. Theoretically, this would block IL-12 functionality, unless the linker was cleaved. This construct, named “pro-IL-12,” indeed had reduced bioactivity, which was restored after incubation with MMP14 and was dependent on the presence of the cleavable linker.
Xue and Moon et al. next characterized how pro-IL-12 behaved in vivo. The engineered IL-12 extended survival in mice bearing MC38, B16F10, or 4T1 tumors, comparable to the non-masked hetero-IL-12–Fc. However, relative to hetero-IL-12–Fc, pro-IL-12 reduced serum cytokine levels, weight loss, and liver damage, providing the same therapeutic benefit with a clear improvement of toxicity.
Next, the researchers questioned which cell types could be responding to pro-IL-12. In a series of depletion experiments, they found that NK cells, neutrophils, and macrophages were not necessary for pro-IL-12’s efficacy, suggesting that innate immune cells are not the key responders. On the other hand, pro-IL-12 was ineffective in Rag1-/- mice, or mice depleted of CD8+ (but not CD4+) T cells, pointing to CD8+ T cells as the target. The likely responders were more specifically CD8+ T cells within the TME, as pro-IL-12 was still effective in mice treated with the lymphocyte egress inhibitor FTY720 (which would prevent migration from lymphoid organs to the tumor). The team found that pro-IL-12 increased tumor antigen-specific and IFNγ-expressing CD8+ T cells, and decreased exhaustion markers such as PD-1 and Tim3. Interestingly, co-administration of an anti-IFNγ antibody abrogated the therapeutic effect, demonstrating that the therapeutic response to pro-IL-12 involves IFNγ and CD8+ T cells.
The team next investigated the specific cellular source of IFNγ. Bulk splenocytes were treated with hetero-IL-12-Fc ex vivo, stimulating IFNγ production as expected. Consistent with the in vivo depletion results, depletion of NK cells from the splenocytes did not reduce IFNγ production, while loss of CD4+ T cells led to a slight decrease, and loss of CD8+ T cells led to a substantial decrease, again highlighting CD8+ T cells as the main responders. To test whether CD8+ T cells bound IL-12 themselves, the researchers co-cultured mixtures of CD4+ and CD8+ T cells taken either from WT or IL-12R-/- (IL-12 non-responsive) mice. Interestingly, WT CD8+ T cells mixed with IL-12R-/- CD4+ T cells and stimulated with hetero-IL-12-Fc had comparable IFNγ production to the fully WT control, while WT CD4+ T cells mixed with IL-12R-/- CD8+ T cells did not secrete IFNγ. These results were substantiated in vivo. When the cell mixtures were transferred into Rag1-/- mice bearing MC38 tumors, only the mice with IL-12R-competent CD8+ T cells responded to pro-IL-12 therapy. Altogether, these results suggested that CD8+ T cells directly sense IL-12, triggering IFNγ production.
Given the ability of pro-IL-12 to activate IFNγ production in the TME, the researchers tested whether pro-IL-12 could supplement other cancer therapies. In the TUBO HER2+ breast cancer model, pro-IL-12 along with an EGFR tyrosine kinase inhibitor (TKI) substantially improved tumor control and survival over pro-IL-12 or TKI alone. Furthermore, the combination of pro-IL-12 and anti-PD-L1 reduced tumor growth and improved survival in MC38-bearing mice – again better than either therapy alone – indicating pro-IL-12’s potential in combination therapies to activate the TME.
Finally, the team considered the application of this approach to human IL-12. They constructed a human hetero-IL-12–Fc, which induced IFNγ production from human PBMCs, and developed a prodrug version that became activated after MMP14 exposure. In a humanized mouse model, the human IL-12 prodrug slowed growth of a human colon cancer cell line, with reduced toxicity compared to the hetero-IL-12–Fc version. These results suggested potential for this same therapeutic strategy in human cancer patients.
Altogether, Xue, Moon, and the team engineered a more stable and less toxic IL-12 that could regress tumors and synergize with other cancer therapies. The approach to make this cytokine fit for use could be extended to a variety of other cytokines and chemokines to support antitumor efficacy. More broadly, the utility of a strategy that can heat up the TME may support a variety of combination therapies in treating diverse cancers.
Write-up by Alex Najibi, image by Lauren Hitchings



