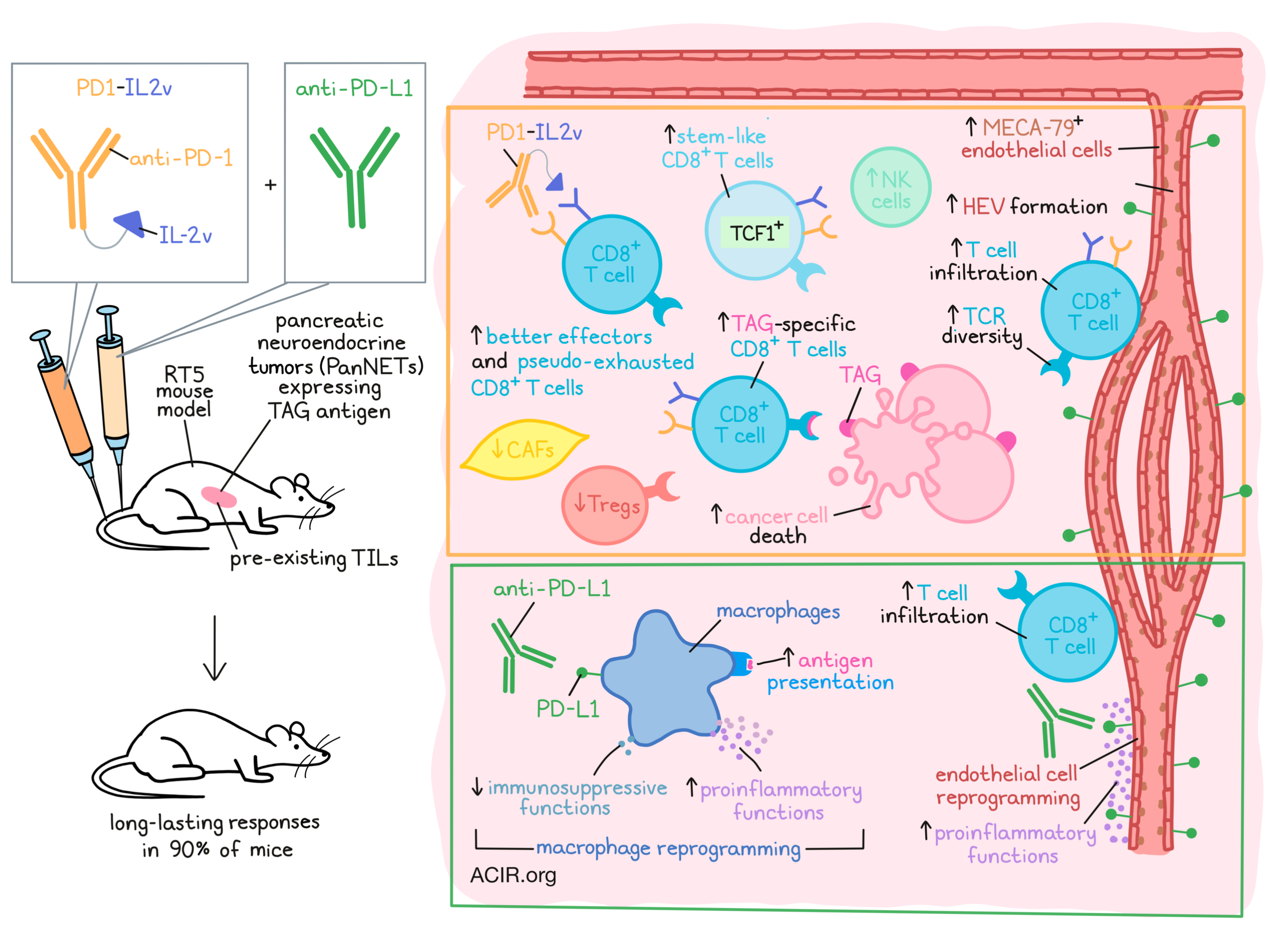
The immunocytokine PD1-IL2v, a fusion of an anti-PD-1 antibody with an IL-2 variant that does not stimulate Treg responses, has been developed to target activation of CD8+ T cells in the tumor via PD-1 binding. Tichet and Wullschleger et al. investigated mechanisms behind intrinsic and adaptive resistance to this type of therapy in a spontaneous mouse model for pancreatic cancer. Their research was recently published in Immunity.
The researchers used the RT5 spontaneous mouse model for pancreatic cancer, in which transgenic mice express a driving oncogene encoding the SV40 large T-antigen (TAG) oncoprotein. RT5 mice develop a spontaneous immune response against TAG, resulting in TIL in premalignant lesions. Spontaneous pancreatic neuroendocrine tumors (PanNETs) that develop in RT5 mice avoid immune destruction and form solid tumors that are resistant to immunotherapy.
Tichet and Wullschleger et al. began by conducting a pharmacodynamic study, choosing a dose of PD1-IL2v that does not block all PD-1 molecules on T cells, but instead would deliver sufficient IL-2v to the T cells in the tumor microenvironment (TME) and lymphoid organs to be efficacious, but non-toxic. Assessing different treatment groups, including PD1-IL2v and IL-2v or anti-PD-1 monotherapy, groups receiving IL-2v showed a 3-4x increase in CD8+ T cells in the spleen. However, only the PD1-IL2v group had an increase in TAG-specific CD8+ T cells. No increases in Tregs were observed, as has been shown in prior IL-2v treatments. In tumors, there was also a clear increase in CD8+ T cell infiltration after PD1-IL2v treatment; both the total and TAG-specific CD8+ T cell counts went up 10-fold, and these cells expressed PD-1. Treatment induced a significant increase in stem-like PD-1+TCF-1+CD8+ T cells and their progeny, PD-1+TCF-1-CD8+ T cells. Looking at activation markers, PD1-IL2v induced PD-1+TCF-1-CD44+ effector CD8+ T cells, which expressed granzyme B. More specifically, these were “fresh” effector (PD-1+CD218a+) and “better” effector (PD-1+TCF-1-CD218a+Tim-3hi) CD8+ T cells. Vascularity also changed after treatment, with PD1-IL2v inducing MECA-79+ endothelial cells that had features of high endothelial venules (HEVs).
To assess therapeutic efficacy against established, autochthonous tumors, mice began treatment once tumors reached approximately 10 mm3 volume and were monitored after treatment by ultrasound imaging for 16 weeks. Untreated tumors kept progressing, but PD1-IL2v resulted in tumor regression in most mice, with 40% of mice having complete responses. Tumors that relapsed after initial regression had limited numbers of TAG-specific CD8+ T cells, and HEVs were similar to untreated tumors. Depletion and antibody blockade experiments showed that the therapeutic effects of PD1-IL2v were dependent on CD8+ T cells and IFNγ signaling. In tumors that developed acquired resistance to treatment, PD-L1 was upregulated, most prominently on the tumor vasculature and on macrophages in the TME.
To address this acquired resistance mechanism, the researchers then assessed whether combining the treatment with PD-L1 blockade could overcome resistance. Tumors regressed rapidly in the first four weeks of combination treatment, resulting in long-lasting responses without relapses in 90% of mice.
To uncover the underlying molecular mechanisms driving the differences in therapeutic efficacy, the tumors were examined during the early phase of treatment, when maximal tumor regression was taking place. Tumors and spleens were collected after 10 days and subjected to single-cell RNAseq and scTCRseq. In spleens, there was a 2-fold increase in CD8+ T and NK cells in both treatment groups, with a particular increase in effector memory CD8+ T cells after PD1-IL2v treatment. In the tumor, PD1-IL2v induced strong T cell infiltration, and combining it with anti-PD-L1 did not change cell type abundances in the TME. Furthermore, in both groups (PD1-IL2v w/ or w/o anti-PD-L1), the percentage of macrophages and cancer-associated fibroblasts (CAFs) was reduced.
Untreated tumors were significantly infiltrated with naive T cells, and 14% of the TILs had an effector memory (TEM) phenotype. Tumors treated with PD1-IL2v were abundant in TEM and exhausted (TEX) CD8+ T cells, while Tregs were decreased. No changes were observed in the phenotype of CD8+ T cells when anti-PD-L1 was added to PD1-IL2v.
Assessing the functional states of CD8+ T cells, the researchers found that TEM CD8+ cells had IL2-Stat5 and TNF-A signaling pathway enrichment, indicative of “better effectors”. These cells also had higher expression of genes involved in T cell activation and cytotoxicity. In TEX, there were also increases in the expression of Gzmba, Tnf, and Ctla2a, and TNF-A and IL2-Stat5 signaling pathways were also enriched. This indicates that these exhausted T cells were not terminally exhausted, but “pseudo-exhausted”.
Next, the researchers investigated the TCR repertoire of TIL after treatment. The richness of the TCR repertoire was similar in the treatment groups, but the total number of clones in tumors treated with PD1-IL2v or combination therapy was higher than those treated with anti-PD-L1 alone. In the PD1-IL2v groups, over 50% of T cell clones were expanded and found over 10 times. Expanded clones were associated with the “better effector” TEM and “pseudo-exhausted” TEX CD8+ phenotypes.
To more comprehensively assess the effects of combination with PD-L1 blockade, the researchers turned to an evaluation of myeloid cells in the tumor. Ex vivo assays showed that tumor-derived myeloid cells were immunosuppressive. In untreated tumors, an M2-like Mrc1+ tumor-associated macrophage (TAM) population was found that expressed the immunosuppressive cytokine IL-10. scRNAseq of myeloid cells showed that PD-L1 blockade impacted the phenotype and function of TAMs, polarizing these cells toward a pro-inflammatory and APC-like phenotype, while there was a reduction of genes related to M2-like immunosuppression. When comparing the combination treatment with PD1-IL2v alone, there was an increase in IL-6, TNFα, and inflammatory signaling pathways in macrophage and DC clusters when PD-L1 blockade was added. These data suggest that the addition of anti-PD-L1 to PD1-IL2v resulted in a switch from immunosuppressive TAMs to a more pro-inflammatory and antigen-presentation phenotype. Furthermore, combination treatment also impacted the phenotype and function of the vasculature in the tumor, leading to more effective T cell infiltration.
Finally, the researchers confirmed all of the effects in the syngeneic GL261 orthotopic transplant glioma model. Overall, this preclinical work shows that the efficacy of PD1-IL2v treatment may be improved by adding PD-L1 blockade, which aids the T cell responses induced by PD1-IL2v treatment by remodeling the tumor microenvironment into a more pro-inflammatory environment, allowing complete and sustained tumor remissions. These translational studies demonstrate the potential benefit of therapy with this novel immunocytokine, particularly in patients who have developed acquired resistance to anti-PD-1 axis therapy but who still exhibit infiltrating T cells.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, co-first authors Mélanie Tichet and Stephan Wullschleger answered our questions.

What was the most surprising finding of this study for you?
SW and MT: Our lab focuses on the tumor microenvironment (TME) and understanding resistance to therapies using various mouse models. We took advantage of the RIP1-Tag5 (RT5) model established by Doug in 1984; indeed, this spontaneous cancer model of pancreatic neuroendocrine tumor, infiltrated with inactive tumor-specific T cells, is resistant to classical immune checkpoint blockers (ICB), such as anti-PD-1. We found that PD1-IL2v monotherapy specifically stimulates PD-1+ stem-like tumor-specific CD8+ T cells leading to cancer cell killing and therapeutic benefit; however, the response was transitory. We then combined PD1-IL2v with anti-PD-L1, which blocks the immune checkpoint ligand PD-L1, which resulted in significantly improved tumor shrinkage and survival benefit. But, to our surprise, anti-PD-L1 did not directly alter the activity of T cells as expected for a checkpoint inhibitor; rather, it remodeled components of an immunosuppressive barrier in the TME to adopt pro-inflammatory phenotypes: macrophages and the tumor vasculature.
What is the outlook?
SW and MT: It will be important to further investigate the efficacy of PD1-IL2v plus anti-PD-L1 in different tumor types. As a first step, we have demonstrated the benefits of this combination therapy in a glioblastoma (GBM) model. Additionally, we aim to expand these studies to mechanistically decipher the effects on T cells in lymph nodes vs. the tumor microenvironment. We believe that our studies encourage the clinical development of PD1-IL2v as a new form of immunotherapy. Our collaborators at Roche have launched a clinical trial of PD1-IL2v with and without anti-PD-L1 in solid tumors (NCT04303858); we are eagerly looking forward to the first clinical results of this new therapeutic modality.
What was the coolest thing you’ve learned (about) recently outside of work?
MT: I took advantage of Switzerland's beautiful wilderness and rediscovered nature; the mountains here are the best places to spend time outside the lab. I started skiing again (after a 20 years break!) and discovered ski touring, climbing, and Via Ferrata, and also hiking and mushroom “hunting”. And using these mushrooms afterward to make the most amazing chanterelle/burrata pizza or a porcini risotto is definitely a plus!



