Aiming to uncover the role that the expression of PD-1 in various cell types plays in antitumor immunity, Strauss et al. analyzed the effects of selective PD-1 ablation in myeloid cells and T cells, and surprisingly found that knockout of PD-1 in myeloid cells, but not in T cells, was crucial for an antitumor response. The results of this study were recently published in Science Immunology.
Selecting the B16F10 melanoma model for its known resistance to checkpoint blockade, the researchers started by showing that compared with non-tumor-bearing mice, mice with B16F10 tumors had upregulated expression of PD-1 and PD-L1 on myeloid cells within the spleen and tumor. All subsets of myeloid cells – including monocytic myeloid-derived suppressor cells (M-MDSCs), poly-morphonuclear MDSCs (PMN-MDSCs), CD11b+F4/80+ macrophages, and CD11c+MHC-II+ DCs – expressed PD-1, and levels of PD-1 expression increased over time.
Strauss et al. then examined the myeloid progenitors within the bone marrow; these cells give rise to MDSCs and tumor-associated macrophages during emergency myelopoiesis – a rapid increase in myeloid cell output in response to immunological stress, including tumor formation. The researchers observed low PD-1 expression in granulocyte/macrophage progenitors (GMPs) in non-tumor-bearing mice and elevated PD-1 levels in GMPs and in their precursors, common-myeloid progenitors (CMPs), in tumor-bearing mice. PD-1 levels in myeloid progenitors peaked soon after tumor inoculation, suggesting that an increase in PD-1 expression happens early during tumorigenesis. PD-L1 was constitutively expressed on myeloid progenitors in non-tumor-bearing mice and was elevated in tumor-bearing mice. Furthermore, culture experiments showed that the upregulation of PD-1 on myeloid progenitors from tumor-bearing mice was regulated by growth factors that drive emergency myelopoiesis in tumors.
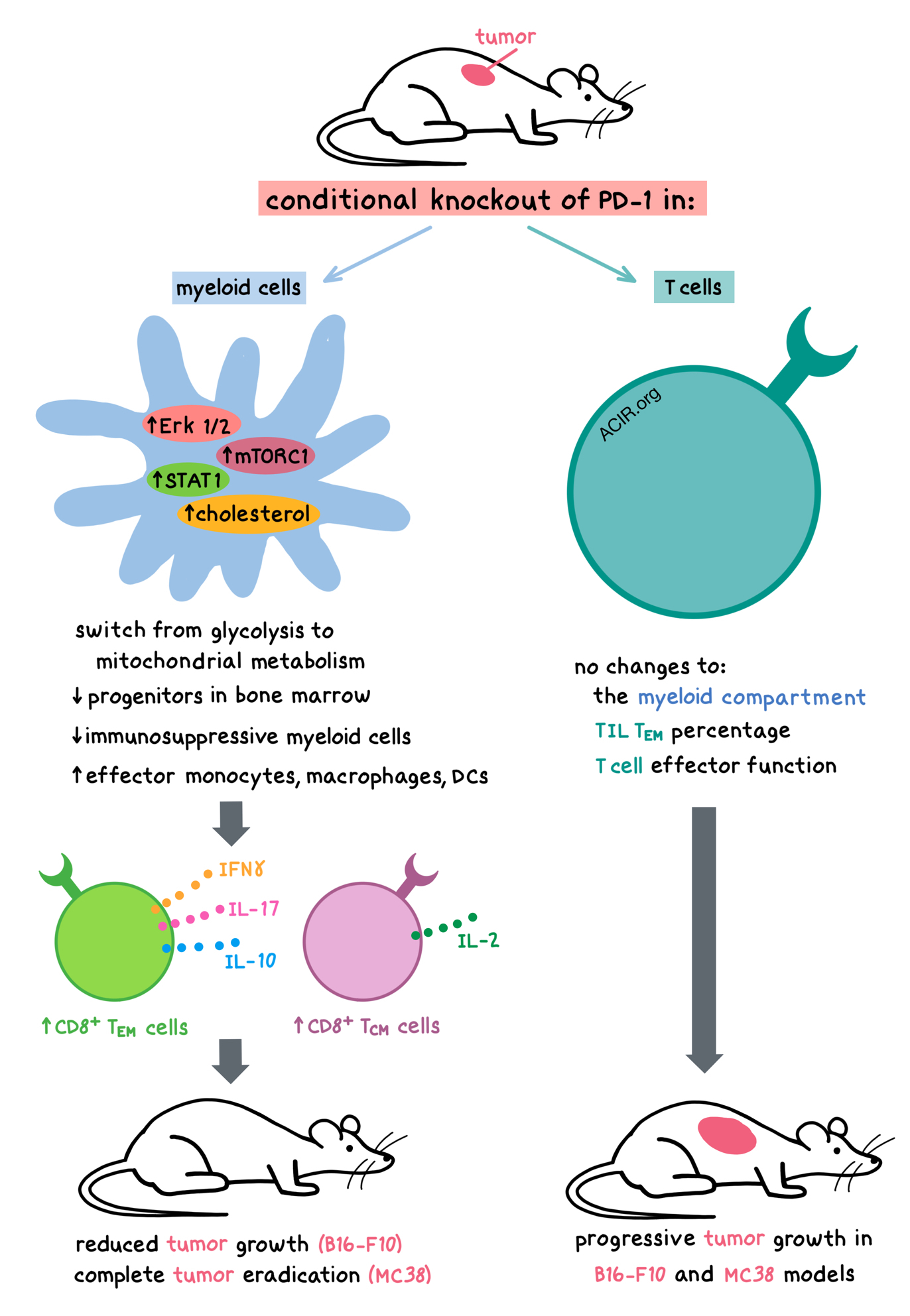
Next, the researchers showed that complete knockout of PD-1 in mice decreased tumor growth and reduced the accumulation of GMPs (but not CMPs) in the bone marrow – GMPs first rapidly expanded, but then declined. PD-1 knockout led myeloid cell differentiation away from immunosuppressive MDSCs and toward effector monocytes, macrophages, and DCs in the tumor, spleen, and small intestine. These effects on the myeloid compartment in the PD-1-/- mice were seen even before tumors became measurable, and were observed in two other murine tumor models. Importantly, similar effects on myeloid cells were observed with PD-1 blockade using anti-PD-1 antibody.
To tease out the effects of PD-1 ablation within specific cell populations, Strauss et al. created mice with a conditional PD-1 knockout in either the myeloid cells (conditional ablation of LysM) or in T cells (conditional ablation of CD4). In mice with myeloid-specific PD-1 knockout, B16F10 tumor growth was reduced despite PD-1 expression on T cells. GMP accumulation in the bone marrow was significantly reduced, and levels of immunosuppressive myeloid cells were diminished, while levels of functional effector monocytes, macrophages, and DCs were increased in the tumor and spleen; all of these phenotypes were also observed in complete PD-1 knockouts. In contrast, T cell-specific PD-1 knockout did not alter the myeloid compartment, the percentage of tumor-infiltrating effector memory T cells, or T cell effector function. Thus, T cell-specific PD-1 knockout did not provide as much antitumor protection as the myeloid-specific PD-1 knockout. Similar results were observed in the highly immunogenic MC38 colon adenocarcinoma model, where myeloid-specific PD-1 knockout led to complete tumor eradication, while T cell-specific PD-1 knockout resulted in progressive tumor growth. Together, these results demonstrate that PD-1 expression on myeloid cells plays a critical role that impacts T cell function by preventing the differentiation of effector myeloid cells and promoting the formation of MDSCs. Therefore, blocking PD-1 signaling in myeloid cells appears to be a requirement for antitumor immunity.
Next, Strauss et al. looked into the mechanisms behind the effects of myeloid-specific PD-1 knockout. In vitro, PD-1-deficient GMPs cultured with granulocyte colony-stimulating factor (G-CSF) showed enhanced activation of Erk1/2, mTORC1, and STAT1 – signaling targets that play a role in the differentiation and maturation of myeloid cells and prevention of MDSC generation. Metabolically, PD-1 deficiency transitioned the myeloid precursors from glycolysis to mitochondrial metabolism, which is associated with differentiation. Enhanced mitochondrial metabolism was also observed in monocytes, macrophages, and DCs isolated from PD-1-knockout mice and myeloid-specific PD-1-knockout mice. Compared with controls, PD-1-deficient myeloid precursors had elevated metabolic intermediates, with the most prominent difference in the level of cholesterol, which is required for the differentiation of proinflammatory monocytes, macrophages, and DCs, and also promotes antigen-presenting function. Metabolism-driven differentiation was observed early in tumorigenesis in PD-1-knockout mice.
In addition to affecting the myeloid cells directly, myeloid-specific PD-1 knockout also affected T cells. Tumor-bearing mice with myeloid-specific PD-1 knockout had more IFNγ-, IL-17-, and IL-10-producing CD8+ effector memory T cells, as well as more IL-2-producing CD8+ central memory T cells compared to control tumor-bearing mice. CD8+ T cells in tumor-bearing mice with myeloid-specific PD-1 knockout had elevated levels of ICOS and Lag3, but not Tim3, CTLA-4, PD-1, PD-L1, or CD160. Furthermore, DCs isolated from spleens of PD-1-knockout tumor-bearing mice were better able to induce antigen-specific CD4+ and CD8+ T cell proliferation and IFNγ expression compared to control tumor-bearing mice.
Overall, the results of this study show that PD-1 knockout in myeloid cells leads to improved systemic antitumor immunity by reducing MDSCs and promoting differentiation of proinflammatory and effector monocytes, macrophages, and DCs, thus resulting in enhanced effector T cell responses and improved tumor control, suggesting that this may be a key mechanism underpinning PD-1 blockade.
by Anna Scherer
Meet the researcher
This week, first author Laura Strauss answered our questions.

What prompted you to tackle this research question?
Immune checkpoint inhibitors (ICI) used for cancer immunotherapy were shown to boost the existing antitumor immune response by preventing the inhibition of T cells by tumor cells. However, 60-70% of cancer patients do not respond to this treatment possibly due to profound systemic and local immunosuppression, which is mediated partly by myeloid cells. Tumor-associated macrophages and their impact on the metabolic environment of the tumor microenvironment (TME) have a major impact on resistance to therapy and metabolic networks. Moreover, accumulating evidence demonstrates that the frequency and immunosuppressive function of myeloid-derived suppressor cells (MDSC) in cancer patients is a strong predictive marker for therapy response and can contribute to patient resistance to immune checkpoint inhibition. Therefore, targeting pathways that fuel the production and expansion of MDSCs from myeloid progenitors might provide novel key targets for solving resistance to immunotherapy, including ICI. We investigated how PD-1 might regulate the production of myeloid cells and metabolic reprogramming of myeloid progenitors and effector myeloid cells in mouse models of cancer.
What was the most surprising finding of this study for you?
We found that mice with conditioned deletion of Pdcd1 in myeloid progenitors and myeloid cells showed a marked change in tumor-driven myelopoiesis, involving a shift in myeloid cell fate away from immature MDSC and towards differentiated monocytes, macrophages, and dendritic cells. Surprisingly, myeloid-specific targeting of Pdcd1 led to more efficient inhibition of tumor growth than T cell-specific Pdcd1 ablation. Further, in immunogenic mouse models, myeloid cell-specific Pdcd1 ablation resulted in complete tumor eradication, whereas T cell-specific Pdcd1 ablation allowed progressive tumor growth.
What was the coolest thing you’ve learned (about) recently outside of work?
I have learned some novel aspects that are important to move your research forward, to stay highly motivated, and to reach a healthy life/work balance: keeping a mindset of a child, staying curious, drawing a sharp boundary between work and home, reserving some hours for “Me-Time”. Having time for ourselves reboots our brains, enhances our relationships, and makes us more productive.



