To gain insight into the mechanisms underlying response and resistance to immunotherapy, Li, van der Leun, Yofe, and Lubling et al. set out to molecularly characterize the expressed gene sets of tumor-infiltrating immune cells in order to quantitatively and qualitatively analyze the heterogeneity within and between cancer patients. The results were recently published in Cell.
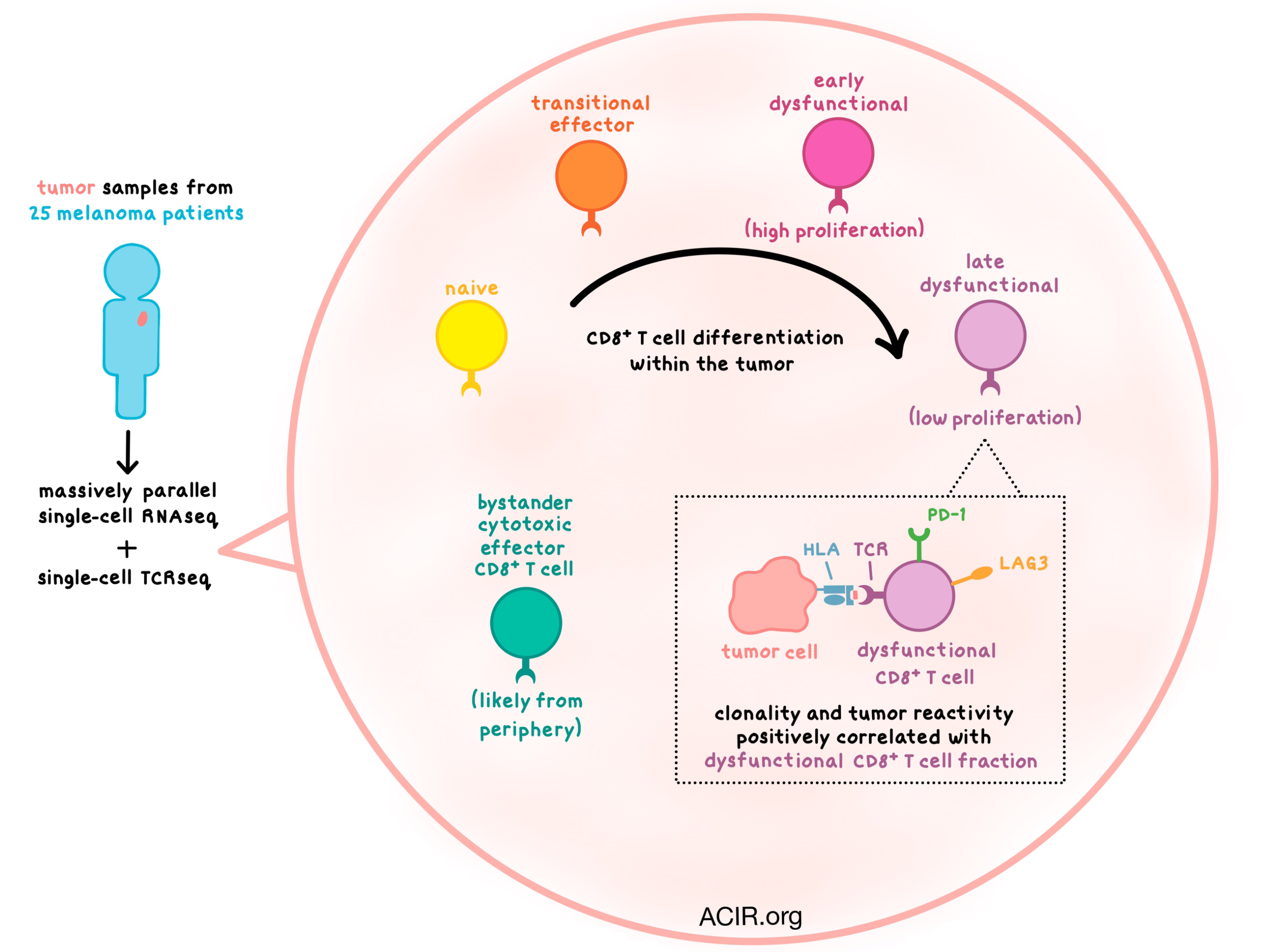
Using massively parallel single-cell RNA sequencing (MARS-seq), the researchers began by examining tumor samples from 25 melanoma patients with various stages of disease and treatment histories. Focusing on T and NK cells first, clustering analysis revealed a number of subsets, including naive T cells; cytotoxic effector (GZMH+), transitional effector (GZMK+), and dysfunctional (PD-1+LAG3+) CD8+ T cells; a small subset of CD4+ and CD8+ memory T cells; Tregs, follicular helper (TFH), and dysfunctional CD4+ T cells; and NK cells. The largest CD8+ T cell cluster was the dysfunctional subset. The observed heterogeneity of molecular phenotypes among T cells was consistent across patients with different disease stages, metastatic sites, and prior treatments, pointing to a universal process that shapes the T cell population within melanoma tumors.
The researchers then used the MARS-seq data to create two transcriptional scores: one for dysfunction (including LAG3, PD-1, and CXCL13) and one for cytotoxicity (including FGFBP2 and CX3CR1). Based on these scores and the clustering analysis, they observed a spectrum of CD8+ T cell states, on a continuum from naive, to transitional, to dysfunctional, indicating that these cells gradually increased their expression of immune checkpoint genes. A similar gradient was observed with Tregs, and interestingly there was some overlap in genes and transcription factors activated in both Tregs and dysfunctional CD8+ T cells. However, the cytotoxic T cells appeared to form a distinct cell population.
Turning their analysis to myeloid cell subsets, the team identified macrophages, monocytes, dendritic cells, plasmacytoid DCs, osteoclast-like cells, B cells, and plasma cells. The data suggested that, similar to T cells, monocytes undergo continuous development and differentiation within the tumor, and that the tumor is not just infiltrated with mature myeloid cells.
Comparing data between patients, the researchers saw that while the myeloid cell distribution was patient-specific, most of the T cell transcriptional states showed up in many patients (albeit with varying frequency). The dysfunctional CD8+ T cell population fraction was particularly variable (ranging from 3.6% to 72.1% of tumor-infiltrating T cells), and it was negatively correlated with naive-like T cell fraction and positively correlated with TFH fraction.
To gain a better understanding of the clonal structure and dynamics of various T cell states, the researchers coupled MARS-seq with single-cell TCRβ sequencing. TCR clonotype distribution varied widely across patients, with some T cell infiltrates exhibiting large TCR diversity but little clonal expansion, and with other T cell infiltrates dominated by a few T cell clones. Clonality positively correlated with the dysfunctional CD8+ T cell fraction, which constituted the most proliferative tumor-infiltrating immune cell subset. More proliferation was observed in the early stages of the dysfunctional program acquisition, but with increased dysfunctionality, proliferation tapered off. Similarly, Tregs were highly proliferative mostly in the early stages of differentiation.
TCRβ genotype provided an opportunity to link clonality and phenotype. Clonal analysis once again showed overlap between transitional and dysfunctional CD8+ T cells, while cytotoxic cells formed a distinct “bystander” subset. To find a potential source of these cytotoxic T cells, the team conducted MARS-seq of peripheral blood, and found that PBMCs contained no dysfunctional cells, but had naive-like, transitional, and cytotoxic CD8+ T cells. This suggests that cytotoxic cells arrive from the periphery rather than differentiate within the tumor.
Asking the question of whether their study results would extend beyond melanoma, the researchers analyzed a dataset of T cells from lung adenocarcinoma samples. They found some differences in the specifics of the dysfunctional gene program between tumor types, but most observations were consistent with melanoma data.
Finally, the team analyzed the relationship between T cell states and tumor reactivity by co-culturing ex vivo-expanded patient T cells with autologous tumor cells. Tumor reactivity, measured by IFNγ and TNFα production, varied significantly between TILs from different patients, with some TILs showing no reactivity. In general, tumor-reactive T cells tended to be in a dysfunctional CD8+ T cell state with a trend toward increasing reactivity with increasing dysfunctionality.
Taken together, the transcription, clonality, and proliferation data suggest a model in which antigen-driven interactions within the tumor microenvironment lead to continuous T cell differentiation toward an early dysfunctional and then highly dysfunctional state. Although the tumor reactivity of the cytotoxic T cells could not be directly evaluated, this model, coupled with the albeit weak association between dysfunctionality and tumor reactivity, may suggest that T cells with a cytotoxic signature are bystanders with no relationship to the tumor. Such observations underscore the power of single-cell analysis in unraveling the complexity of immune cell heterogeneity and its relationship to immune cell functionality.
by Anna Scherer



