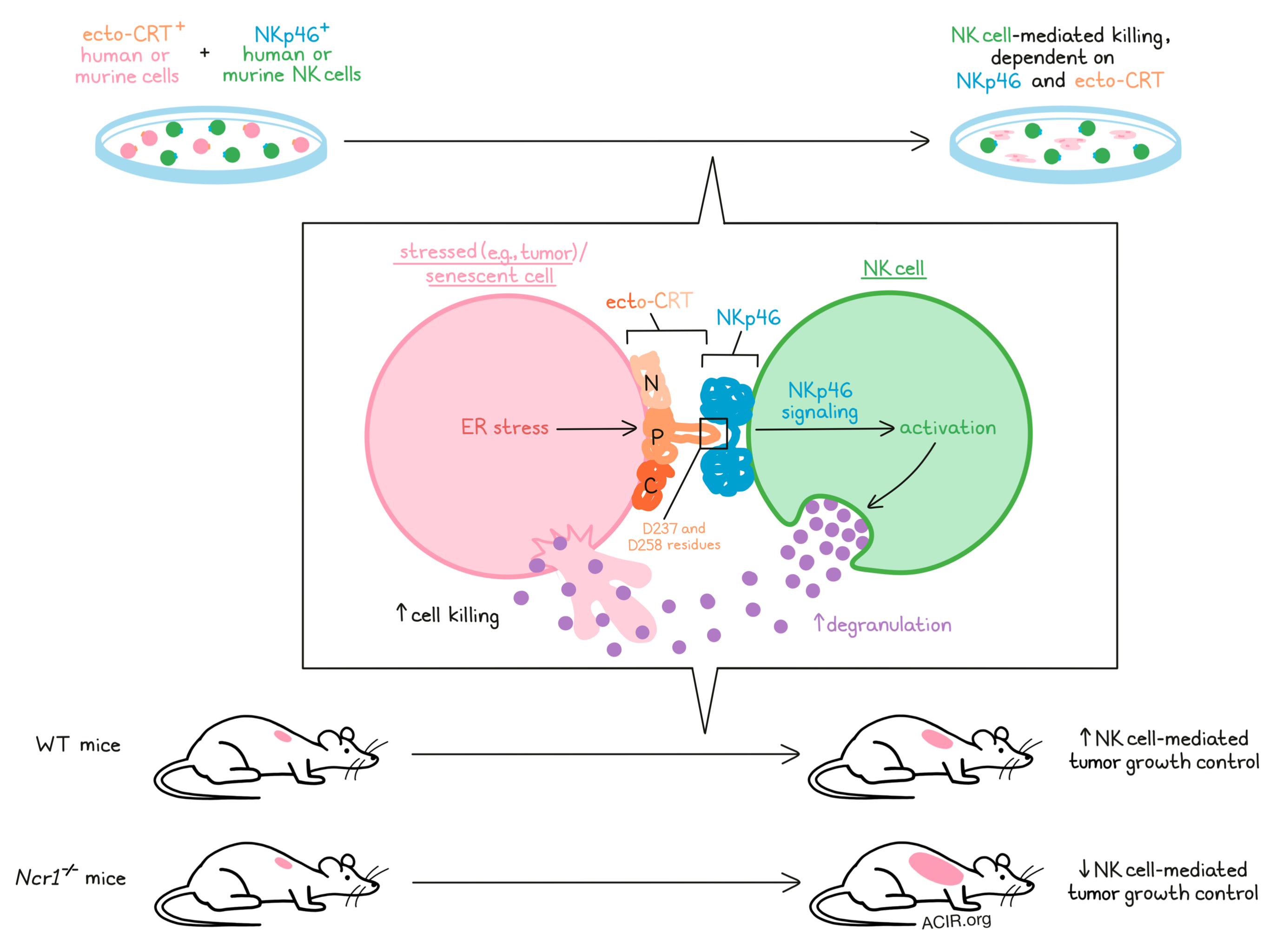
Natural killer (NK) cells are known for their ability to recognize and kill targets that are transformed, infected, stressed out, or senescent, and while NK cells are known to act through several NK cell receptors, many of the ligands that activate those receptors have remained elusive. In a recent study published in Nature, Sen Santara and Lee et al. identified ecto-calreticulin (CRT), a product of ER stress, as an activating ligand of NKp46 that contributes to NK cell-mediated killing of stressed and senescent cells.
To begin, Sen Santara and Lee et al. cultured human peripheral blood NK cells with human JEG-3 choriocarcinoma cells. When JEG-3 cells were infected with ZIKV, which replicates in the ER and causes ER stress, NK cell degranulation and JEG-3 killing were enhanced. This effect was abrogated with the addition of ER stress inhibitors, and was not observed with infections that did not induce ER stress. To identify the NK cell receptor(s) that recognized ER stress, NKp46, NKp30, NKp80, NKG2D, DNAM-1, and 2B4 were each blocked, but only the blockade of NKp46 strongly inhibited NK cell killing of ER-stressed target cells. This effect was confirmed using NCR1 (encoding NKp46) knockout models, and with specific binding of NKp46–Ig (NKp46 fused to the Fc region of human IgG1) to ZIKV-infected cells.
Next, the researchers used NKp46–Ig and NKG2D–Ig to pull down cross-linked ligands from ZIKV-infected JEG-3 cell membranes, and found that the NKp46–Ig alone pulled down a band containing calreticulin (CRT) and protein disulfide isomerases (PDI; PDIA1, and PDIA3), which are known to selectively translocate and associate on the cell surface during ER stress. Increased expression of CRT and PDI was confirmed in JEG-3 cells infected with ZIKV or treated with tunicamycin (an ER stressor). Increased NKp46–Ig binding to ecto-CRT was also observed in JEG-3 cells treated with oxaliplatin (an immunogenic cell death [ICD]-inducing chemotherapy). Importantly, the researchers also showed that binding between ecto-CRT on oxaliplatin-treated cells and NKp46 on NK cells induced downstream NKp46 signaling – an effect that was lost with NKp46 knockout or CRT blockade – and mediating NK cell killing.
Digging deeper into this interaction, the researchers evaluated the three domains that make up CRT (N, P, and C), and showed that NKp46 binds specifically to the P domain. Modeling of CRT binding to NKp46 suggested that the P domain, which forms a hairpin structure, may insert itself into a hinge region between the two immunoglobulins that make up NKp46, forming salt bridges. Using cells expressing mutant forms of CRT, the researchers specifically identified the residues D237 and D258 within the P domain as being essential to NKp46–ecto-CRT binding and subsequent effects.
Parsing out the roles of CRT and PDI, the researchers used small interfering RNA to knockdown CALR and PDIA3. While CALR knockdown reduced ecto-CRT on ZIKV-infected JEG-3 cells by about 65%, PDIA3 knockdown reduced ecto-CRT by only 20%. Further, only CALR knockdown reduced NKp46–Ig binding to and killing of ZIKV-infected cells. Similar effects were observed using blocking antibodies.
To determine whether their findings in human cells could be replicated in murine cells, Sen Santara and Lee et al. treated B16F10 melanoma cells (B16) with cisplatin (a non-ICD-inducing drug that does not increase ecto-CRT) or oxaliplatin (an ICD-inducing drug that does increase ecto-CRT). Both cisplatin- and oxaliplatin-treated cells showed increased susceptibility to NK cell-mediated killing. Blocking CRT or NKp46 abrogated this effect only in the oxaliplatin-treated cells, while blocking NKG2D abrogated killing only in the cisplatin-treated cells. Similar effects were observed using NKp46 and NKG2D knockout NK cells, suggesting that in murine NK cells, NKp46 recognition of ecto-CRT contributes to killing of cells treated with an ICD-inducing drug, but not a non-ICD-inducing drug.
Next, the researchers developed a line of engineered B16 cells that stably expressed high levels of the CRT ectodomain with a C-terminal GPI anchor. In line with their high expression of ecto-CRT, B16-GPI-CRT cells showed increased conjugation with NK cells, dependent on NKp46, and showed evidence of capping (cross-linking-induced movement of groups of cross-linked molecules to one end of the cell) of NKp46 and the integrin CD49 on NK cells with CRT on the target. In vitro, B16-GPI-CRT cells also showed slightly increased colony formation, migration, and invasiveness compared to WT B16 cells, suggesting that ecto-CRT may support malignancy.
In vivo, B16-EV (control) and B16-GPI-CRT tumors grew similarly in Ncr1−/− mice, while in WT mice, B16-GPI-CRT tumor growth was suppressed. Numbers of tumor-infiltrating NK cells, CD8+ T cells, and TAMs were similar between tumors, but a higher portion of infiltrating NK cells in the WT tumors expressed perforin, IFNγ, and TNF, suggesting increased target recognition. Similar patterns were observed in mice bearing B16 tumors treated with doxorubicin (ICD-inducing). B16-GPI-CRT tumors also developed fewer metastases than control tumors in WT mice – an effect that was dependent on NK cells and NKp46. Interestingly, B16-GPI-CRT tumors developed more metastases than control tumors in the Ncr1-/- mice, possibly due to the enhanced malignancy-associated properties that these cells demonstrated in vitro. Myeloid cell depletion did not affect metastases in WT mice with either tumor type, but did increase metastases from both tumor types in Ncr1-/- mice, suggesting that NKp46-mediated NK cell control may be the dominant antitumor mechanism, while myeloid cell protection compensates in its absence.
Finally, the researchers investigated the role of NKp46–ecto-CRT binding in NK cell detection and destruction of senescent cells. To this end, trametinib (T, a MEK inhibitor) and palbociclib (P, a CDK4/6 inhibitor) (T + P) were used on A549 human lung cancer cells, inducing senescence, ER stress, and ecto-CRT, along with NKG2D ligands. As in previous models, NK cells showed increased killing of T+P-treated cells compared to untreated cells, and blockade of NKp46, CRT, or NKG2D reduced NK cell-mediated killing, with simultaneous blockade of NKp46 and NKG2D reducing killing to background levels. Similar results were observed in mice bearing KP lung tumors, where T+P suppressed tumor growth, dependent on NKp46. While NKG2D also played a role in controlling these tumors, the role of NKp46 was found to be dominant. Similar patterns were observed with other senescence-inducing drugs, depending on whether they induced ER stress.
Together these results show that NK cells recognize stressed and senescent cells through NKp46 binding to ecto-CRT, which is upregulated during ER stress in both human and murine cells. This fundamental biological mechanism is an important step forward in understanding NK cell biology, and will likely be important in understanding and optimizing numerous cancer immunotherapies involving NK cells, and in developing future immunotherapies that could exploit this pathway.
Write-up and image by Lauren Hitchings



