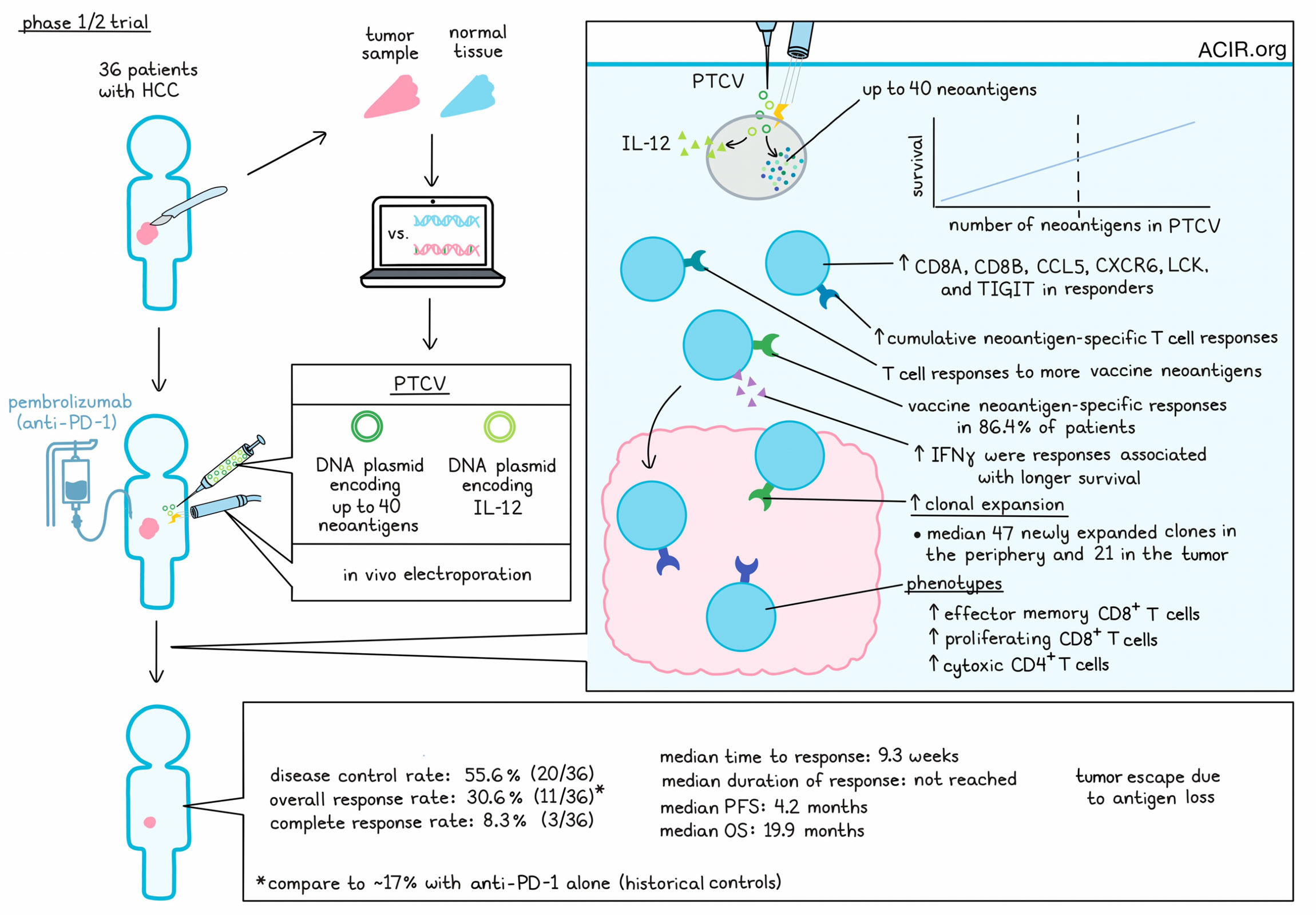
Advanced hepatocellular carcinoma (HCC) is a tumor type with a modest tumor mutation burden (TMB), low T cell infiltration, limited responses to checkpoint inhibitors, and a low patient survival rate. To overcome these hurdles, Yarchoan et al. conducted a single-arm, open-label, phase 1/2 study, in which 36 patients with advanced HCC who had previously been treated with a multityrosine kinase inhibitor (mTKI) were treated with pembrolizumab plus a personalized therapeutic cancer vaccine (PTCV). PTCVs consisted of a DNA plasmid encoding up to 40 personalized neoantigens, coadministered with a second DNA plasmid encoding adjuvant IL-12. PTCV is administered intradermally, followed by in vivo electroporation, resulting in only local and transient IL-12 production.
Vaccine manufacturing was successful and on-schedule for all patients enrolled in the trial. Treatment was safe and well tolerated, with an adverse event profile that was similar to that of pembrolizumab monotherapy in HCC. The most common vaccine-related adverse events were local injection-site reactions.
Among 34 patients who were evaluable for response, the disease control rate was 55.6% (20/36), with an overall response rate (ORR) of 30.6% (11/36), and a complete response rate (CRR) of 8.3% (3/36). This ORR was statistically significant compared to the historical ORR of 17% for pembrolizumab monotherapy in a similar population. Two patients with partial responses saw continued tumor reduction in the target lesion, but new tumor lesions emerged. One patient whose tumor was previously unresectable was able to achieve secondary resectability. The median time to response was 9.3 weeks, The median progression-free survival was 4.2 months, while the median overall survival was 19.9 months. The median duration of response was not reached, and clinical responses were associated with better progression-free and overall survival. Analysis of ctDNA as an additional measure of response generally tracked with MRI results, though 2 patients had ctDNA responses that appeared to outperform their visible responses.
In analyses of biomarkers, clinical responses were not correlated with pre-treatment TMB, CD8+ T cell infiltration, PD-L1, KDR, or a T cell-inflamed gene signature. However, there was a positive correlation between clinical responses and the number of neoantigens included in the PTCV. A median of 30 neoantigens were included in the vaccines, and patients with 30 or more neoantigens had a 41.2% ORR, while patients with less than 30 neoantigens had a 23.5% ORR. There was a significant difference in the number of targeted neoantigens between the CR/PR group and the SD/PD groups.
Looking at biomarkers of T cell activation and infiltration, the researchers noted increased expression of CD8A, CD8B, CCL5, CXCR6, LCK and TIGIT in responders compared to non-responders. These results were consistent with the proposed mechanism of PTCV inducing new antitumor immune responses.
Next, evaluating whether their vaccines induced specific responses to the peptides that had been predicted and included in the vaccine, the researchers found that among 22 evaluable patients, PTCV treatment was almost always associated with an increase in the magnitude of cumulative PTCV neoantigen-specific T cell responses. In 19 of 22 patients (86.4%), treatment was also associated with an increased number of encoded neoantigens eliciting an immune response, with measurable T cell responses to a median of 64% of the included epitopes, up from a median of 11.8% prior to treatment. These effects were observed in both clinically responding and non-responding patients, and across neoepitopes with predicted high, median, and low binding affinity to HLA-I. The researchers noted a positive correlation between the number of neoantigens included in PTCV and the number of neoantigens that elicited responses. Additionally, patients with IFNγ responses in the top quartile trended towards longer OS than those with IFNγ responses in the bottom quartile. Similarly, patients with CR/PR showed a trend towards greater magnitudes of IFNγ responses.
Neoantigen-specific responses were experimentally confirmed in four responding patients by testing PBMCs against neoepitope pools in vitro. Upon neoantigen stimulation, both CD4+ and CD8+ T cell populations presented increased activation profiles, demonstrating proliferative, polyfunctional, and cytolytic capacities.
TCR sequencing from pre- and post-treatment PBMC and tumor samples showed evidence of significant PTCV-induced clonal T cell expansion in both the tumor and peripheral blood of all 14 evaluable patients, with a median of 47 new or expanded clones in the periphery and a median of 21 new or expanded clones in the tumor. T cell clones were also increased in abundance in both locations, with significantly increased clonality in the tumor, and no change in the TCR repertoire richness. Together these results are consistent with PTCV inducing expansion of T cells in the periphery, and T cells trafficking to the tumor.
Looking at the phenotypes of expanded T cell clones, Yarchoan et al. performed paired scRNA/TCRseq in 4 samples from 3 patients and found that clonally expanded T cells were most associated with effector memory CD8+ T cells, proliferating CD8+ T cells, and CD4+ CTLs, all of which expressed high levels of cytotoxicity-associated genes. Finally, in two representative patients, expanded TCR clones from TILs were mapped to vaccine-encoded epitopes, and specific responses to vaccine neoantigens were observed in both CD4+ and CD8+ T cells, validating the increased infiltration of T cells specific for vaccine neoantigens.
Investigating a case of immune escape in which a patient experienced target lesion regression, but relapsed with a new lesion, the researchers evaluated paired tumor biopsy samples. In the treated lesion, they observed increased infiltration of activated antigen-specific CD8+ and CD4+ T cells, with specific responses to 9 vaccine epitopes, and strong responses to 4. The escape lesion shared some neoepitopes with the target lesion, but had lower T cell infiltration, and had lost expression of the 4 epitopes that had induced the strongest responses in the primary lesion, consistent with immune escape through immunoediting.
Overall, this early clinical trial met its primary endpoints of safety and immunogenicity, as well as its secondary endpoints of treatment efficacy and feasibility, with evidence that PTCV broadens tumor-specific responses and improves clinical responses compared to pembrolizumab monotherapy. Further, responses were associated with higher numbers of neoantigens included in the vaccine, which will help to guide PTCV formulations and patient selection in future trials.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Mark Yarchoan answered our questions.

What was the most surprising finding of this study for you?
Creating a personalized cancer vaccine for individual patients was already an ambitious undertaking, but this clinical study faced an additional challenge by commencing just before the world grappled with the COVID-19 pandemic. Here at Johns Hopkins, where I practice, many of our crucial research personnel were reassigned to provide care for COVID-19 inpatients, while our laboratory team was occupied manufacturing COVID-19 testing kits in our GMP facility. Despite this hectic period, I'm proud that we managed to enroll patients. One surprising outcome was that patients who received the vaccine along with pembrolizumab showed a higher response rate than historically observed with pembrolizumab alone in HCC. Perhaps I'm naturally skeptical, but I wasn't initially convinced that the treatment would yield significant responses. The study primarily aimed to assess safety and immunogenicity, with the hope that these findings might translate into longer-term survival benefits that wouldn't be captured by a small study like this; therefore, the response rate data came as an unexpected and wonderful surprise to me.
What is the outlook?
The data support further development in hepatocellular carcinoma (HCC) and expansion into other challenging tumor types where PD1-based immunotherapy has historically shown limited success due to a lack of tumor-infiltrating T cells. Additionally, reliably inducing tumor-directed T cells, especially CD8+ T cells, has been a challenge in cancer vaccination research. In our study, we demonstrated that the PTCV platform can successfully induce and mobilize T cells into the tumor, thus advocating for the utilization of a PTCV-driven T cell potentiation framework. This framework can be utilized to explore combinations with other immune checkpoint agents and tumor microenvironment (TME) modulators beyond PD-1.
What was the coolest thing you’ve learned (about) recently outside of work?
My kids recently received their standard pediatric vaccines. I tried to explain how vaccines prevent illness by targeting “bad guys”. My son, only 5 years old and with a limited understanding of my work, but aware that cancer is harmful, asked, 'Why can't vaccines fight cancer?”. Good ideas are often simple enough for children to grasp, and that moment was an “aha” for me. Why don't we have widely used vaccines for treating and preventing cancer?



