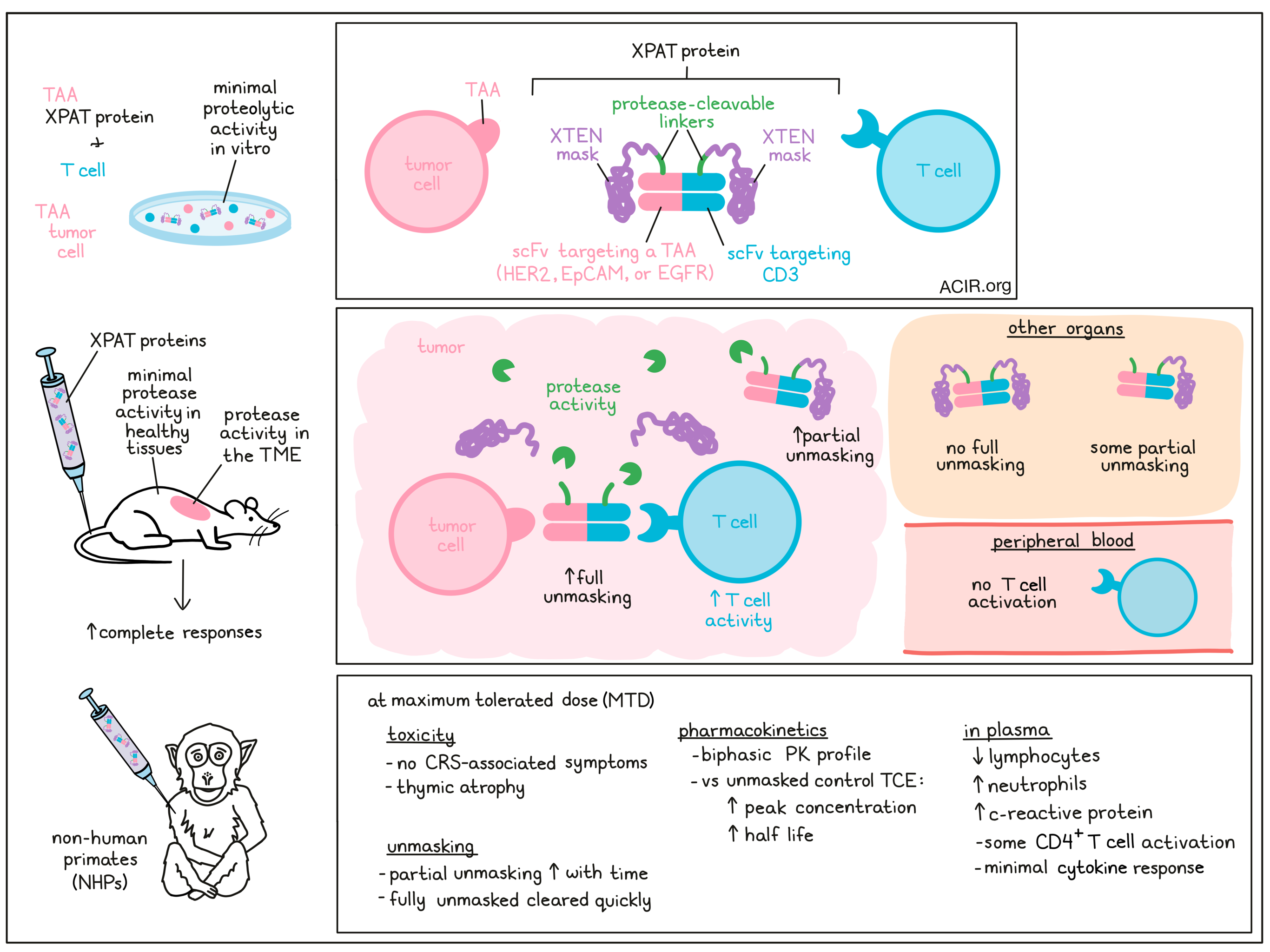
Bispecific T cell engagers (TCE) targeting tumor-associated antigens (TAA) that are expressed at low levels in healthy tissues can cause significant toxicity due to on-target, off-tumor immune effects. To overcome this issue, Cattaruzza et al. developed a protease-releasable masking technology that allows for the creation of precision-activated TCEs (XPAT proteins) that are unmasked by protease activity in the tumor microenvironment. The preclinical research assessing the efficacy and safety of these proteins was recently published in Nature Cancer.
XPAT proteins consist of a TCE core with two “masked” scFvs – one targeting a TAA for specificity, and the other targeting CD3 to activate T cells. The mask is a large, unstructured polypeptide (XTEN) attached to each scFv via a protease-cleavable linker, and blocks the scFv binding site. For this study, the researchers produced XPAT proteins that target the TAAs HER2, EGFR, and EpCAM. Using these proteins, the effects of XTEN polypeptide masking on binding and activity were assessed in vitro. Kinetic analyses of the HER2-XPAT showed that the presence of XTEN masks reduced target affinity 10x for HER2 and 6x for CD3 compared to the unmasked HER2-TCE. In vitro cytotoxicity was measured by coculturing human peripheral blood mononuclear cells (huPBMCs) with the HER2-expressing SKOV3 and BT-474 tumor cell lines. Compared to HER2-TCE, the cytotoxicity was very strongly reduced (multiple log reduction) for the HER2-XPAT protein. This low cytotoxicity was partly due to low proteolytic cleavage during the in vitro assay.
Moving to in vivo models, the researchers evaluated the antitumor activity of the HER2-XPAT protein in HER2hi BT-474 human breast tumors and HER2lo HT-55 colorectal tumors. Tumors were implanted subcutaneously into immunodeficient mice, which were then engrafted with huPBMCs. Both HER2-XPAT and HER2-TCE induced complete tumor regressions within 35 days in the HER2hi model. No activity was observed with a non-cleavable version of HER2-XPAT, indicating that the tumor regression was dependent on protease activity. T cell activity in the tumor microenvironment, measured by CD25 expression on helper and cytotoxic T cells, was similar for both treatments, while T cells were not activated in the peripheral blood (where there was no HER2 expression). In the HER2lo model, there was significant growth inhibition for both HER2-XPAT and HER2-TCE. The body weight of the mice in both models treated with HER2-XPAT remained stable, suggesting limited toxicity.
The researchers created two fluorophore-labeled XPAT proteins to quantify unmasking in vivo in immunodeficient mice with patient-derived xenografts (PDX). Following injection of HER2-XPAT protein, no fully unmasked HER2-TCE and very limited partially unmasked XPAT protein was detected in normal organs. To assess unmasking in tumors, HER2-XPAT and EpCAM-XPAT proteins were used in the relevant PDX tumors. There was and average of 23.8% of unmasked protein among all XPAT proteins across these tumors, and substantial levels of partially unmasked XPAT protein were also present.
Given that the HER2-XPAT protein binds human and non-human primate (NHP) HER2 and CD3 with similar affinity, a dose-escalation study could be conducted in NHP to assess pharmacokinetics (PK), tolerability, and maximum tolerated dose (MTD). Minimal toxicity was detected at doses ≤42 mg/kg (defined as the MTD), with severe toxicity (lethality following a second dose) measured at 50 mg/kg. No cytokine-release syndrome (CRS)-associated symptoms or toxicity was measured at the MTD dose, while low-dose (0.3 mg/kg) unmasked HER2-TCE induced CRS-associated death. The HER2-XPAT protein induced a transient, dose-dependent decrease of lymphocytes, increase in neutrophils, and limited increases in c-reactive protein and CD4+ T cell activation in plasma. At the MTD, the only adverse histopathological event was thymic atrophy.
Plasma concentrations of the HER2-XPAT protein showed a biphasic PK profile, with much higher peak concentrations at the MTD than the unmasked version, and the half-life of the masked protein was 3 days, while it was only 2 hours for the unmasked version in a 3 kg NHP. HER2-XPAT protein induced limited systemic T cell activation and cytokine responses, even at the highest doses tested, with the toxic dose of 50 mg/kg exerting only local toxicity and limited systemic cytokine release.
To assess the in vivo proteolytic stability, the PK profiles of HER2-XPAT protein and HER2-XPAT-NoCLvSite (no cleavage site for proteases) were compared in NHPs. The concentration-time profiles were similar, suggesting the HER2-XPAT protein is stable in vivo, with low cleavage levels in systemic circulation. When high doses were used for treatment, the concentrations of partially unmasked metabolites increased over time, but fully unmasked HER2-TCE remained below the detection level due to rapid clearance.
The stability of the HER2-XPAT protein was tested in spiked plasma samples from humans and NHPs. After a 1-week incubation in human plasma, the protein remained mostly intact, with similar results in samples from patients with cancer or inflammatory diseases and in samples from NHPs (healthy or with systemic inflammation).
Finally, the researchers generated an EGFR-XPAT based on the variable sequences of panitumumab (anti-EGFR antibody) and humanized SP34 (CD3-binding domain). Binding affinity assessment suggested cross-reactivity between EGFR and CD3 from humans and cynomolgus monkeys. There was strong masking of in vitro T cell killing with the EGFR-XPAT protein. In vivo, it had potent antitumor activity against HT-29 human colorectal tumors in huPBMC-engrafted mice. In NHPs, this protein triggered minor toxicity and cytokine release at 0.46 mg/kg. In contrast, the MTD of the unmasked EGFR-TCE was much lower, and resulted in severe CRS-associated toxicity.
These data suggest that the XTEN masking of TCEs can be an effective way to induce tumor regressions, while improving safety when targeting TAAs that are broadly expressed in healthy tissues. The specific activation of the protein in solid tumors allows for higher dosing, potentially improving the efficacy of such treatments in a larger subset of patients. Clinical trials will have to establish whether this reduction in toxicity allows effective dosing that induces tumor regressions.
Write-up by Maartje Wouters, image by Lauren Hitchings.



