
Last week, the ACIR team attended the AACR Annual Meeting 2019 in Atlanta, Georgia. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
T cells in Immune surveillance, Exhaustion, and Therapies
Emerging Immunotherapy Approaches
TME and Tumor Escape
Clinical Trials
T cells in Immune surveillance, Exhaustion, and Therapies
Robert Schreiber’s talk focused on defining the role of MHC-II-restricted neoantigens in tumor vaccine development and the roles of CD4+ T cells in mediating antitumor responses. Schreiber began by describing the application of a newly developed MHC-II prediction algorithm to neoantigens in the T3 sarcoma mouse model, leading to the validation of mutated Integrin B1 (mItgb1) as a major MHC-II neoantigen (see our coverage from the Koch Institute Immune Engineering Symposium 2019). The ectopic expression of either MHC-II-restricted mItgb1 or MHC-I-restricted neoantigen mLama4 in poorly immunogenic KP sarcomas did not sensitize these highly resistant tumors to immune-mediated destruction, but ectopic expression of both neoantigens together increased the immunogenicity of the tumors. This effect was due to enhanced generation of mLama4-specific cytolytic CD8+ T cells mediated by CD4+ T cell help in their priming and maturation. In mice with dual-flanked KP tumors – one expressing both mLama4 and mItgb1 and the other expressing only mLama4 – only the tumor expressing both the MHC-I- and MHC-II-restricted antigens reduced in size following checkpoint blockade. CD4+ and CD8+ T cell levels were also higher in the tumor expressing both types of epitopes. Together, these results demonstrate a critical role for MHC-II neoantigen expression and CD4+ T cell activity at the tumor site for T cell-mediated tumor regression to occur.
In a talk focused on understanding the relationship between checkpoint blockade and the “education” of T cells, John Wherry used deep immune profiling to interrogate the mechanism of anti-PD-1 therapy, starting with its kinetics. Using active proliferation (expression of Ki67) as a marker of response, Wherry found that peak immune responses, driven largely by CD8+ T cells, occurred within a week of initiating anti-PD-1 treatment, with minimal impact at later time points. To better understand the clinical impact of such a rapid immune response, Wherry’s team compared pre-treatment tumor biopsies to post-treatment tumors that had been surgically resected shortly after anti-PD-1 treatment. They found that rapid reinvigoration (but not reprogramming) of the CD8+ T cells and other responding immune cells was short-lived, but strongly associated with antitumor efficacy, with some responding patients showing complete or near complete pathological response by the time of surgery. Reinvigoration of exhausted T cells, however, is not always sufficient for a clinical response. In a first step towards understanding why, Wherry identified a relationship between reinvigoration of exhausted T cells and tumor burden. When the immune response was too low relative to the tumor burden, patients were unlikely to overcome the tumor and derive clinical benefits. Further, this ratio served as a better predictor of clinical benefit than the magnitude of the immune response alone. Resistance that arose in later stage tumors appeared to be more random and utilized genetic events (e.g. β2M loss). Thus, based on this information, Wherry suggested that patients might benefit more from checkpoint blockade therapies if they are administered earlier in the treatment process.
Padmanee Sharma gave examples of how pre-surgical and tissue-based clinical trials, with appropriate analysis of tumor samples prior to and after therapy, and corollary animal studies can provide mechanistic insights and a rationale for how best to proceed with combination checkpoint therapies. After briefly reviewing previously published clinical and animal work on the discovery of the important role that ICOS+CD4+ T cells play during anti-CTLA-4 therapy, she presented newer results from a trial in which metastatic, castration-resistant prostate cancer (mCRPC) patients were treated with ipilimumab after surgical resection. Whole exome sequencing of the tumor was used to identify potential neoantigen-generating mutations, which ranged in number between 0 and 45 for all but one patient, who had over 300mutations. Screening of peripheral blood showed responses to multiple neoantigens after one or two doses of ipilimumab, and patients whose tumors responded to treatment had a longer time to requiring systemic therapy and stronger responses to prostate tumor-associated antigens (PSA, PSMA, PAP). In a neoadjuvant ipilimumab trial, previously “cold” tumors were turned “hot” with multiple immune cell types and biomarkers increasing (or decreasing as appropriate). However, PD-L1 levels also rose on both immune and tumor cells, prompting a follow-on trial in mCRPC with combination ipilimumab and anti-PD-1 therapy, which has just begun. Finally, to understand what options were available for prostate metastases to the bone, Sharma conducted animal studies comparing bone and subcutaneous tumor implants, which showed that bone metastases were resistant to combined checkpoint therapy due to high expression of TGFβ, and that combining checkpoint therapy with blockade of either TGFβ or RANKL (targeting osteoclast activity) was effective in controlling tumor, raising CD4+ T cell levels, and clonally expanding CD8+ T cells, providing a foundation for future clinical studies.
Aiming to understand the mechanisms behind T cell reinvigoration by anti-PD-1, Brian Miller (from Nicholas Haining’s and Arlene Sharpe’s labs) analyzed CD8+ TILs from mice with B16-OVA melanoma and found that the exhausted TILs consisted of two subsets – progenitor exhausted (Tcf1+Tim3-) and terminally exhausted (Tcf1-Tim3+) – and that these two subsets had complementary functions in the antitumor response. The progenitor exhausted TILs had higher proliferative capacity, stronger persistence, and more diverse cytokine production. PD-1 blockade specifically reinvigorated the progenitor exhausted TILs, which then gave rise to the terminally exhausted TILs. The terminally exhausted TILs produced high levels of IFNγ and mediated cytotoxicity against the tumor cells. In metastatic melanoma patients treated with combination anti-CTLA-4 and anti-PD-1, increased frequency of progenitor exhausted CD8+ T cells was associated with longer survival.
According to Steven Rosenberg, the biggest challenge in cancer immunotherapy is developing effective treatments for metastatic, solid epithelial cancers, which result in over 80% of cancer deaths. Rosenberg outlined how adoptive cell transfer relies on T cells’ ability to recognize cancer antigens, which arise when mutations are expressed in proteins, processed intracellularly, and presented as a peptide on MHC. To identify immunogenic mutations and the T cells that recognize them, Rosenberg utilizes a mutated tandem minigene screen with autologous antigen-presenting cells to capture all relevant MHC loci. Because all candidate peptides can be included in this screen, the system is able to identify neoantigen targets that would be missed using neoantigen prediction algorithms. In separate studies of melanoma, gastrointestinal cancers, or epithelial cancers, researchers have used this strategy to identify unique immunogenic neoepitopes, which were rarely shared between patients. Rosenberg and colleagues have begun a series of clinical trials in which the patient-specific neoantigens were identified and neoantigen-targeted T cells were screened and expanded for adoptive transfer. Thus far, the objective response rate in these trials is 15%, though Rosenberg cited two noteworthy patients with dramatic and ongoing complete responses. To improve targeting of somatic mutations in epithelial cancers, Rosenberg showed that the PD-1+ subset of intratumoral T cells have the most potential for tumor recognition, as they were likely activated through prior encounters with tumor antigens. It may also be possible to identify tumor-reactive cells in peripheral blood using 4-1BB as a marker. A combined approach screening both TILs and peripheral blood identified a larger subset of neoantigen-reactive T cells. While most neoantigens are unique, driver mutations, like those in KRAS or p53, may be shared between patients. Rosenberg and colleagues have begun creating libraries of T cell receptors (TCRs) capable of recognizing such shared mutations and suggest that the blueprint for future success will be utilizing both personalized T cells and off-the-shelf neoantigen-targeted T cells selected directly from a library of isolated TCRs.
Crystal Mackall discussed challenges that researchers face in improving the efficacy of CAR T cell therapy, especially given the notoriously high sticker price. Despite a robust level of complete responses, intrinsic or adaptive resistance via reduced or eliminated antigen expression is a significant issue. To this end, Mackall discussed the status of current clinical trials testing a bivalent, CD19/CD22-targeted CAR T cell designed to overcome relapse of antigen-negative or antigen-low tumors. While the trials are ongoing, the bivalent CAR T cells have thus far shown evidence of a strong safety profile and obvious antitumor activity. The second challenge Mackall discussed was CAR T cell failure due to exhaustion. Using a tonic signaling CAR T cell model and ATACseq, Mackall and her team identified regions of chromatin that are differentially accessible in exhausted T cells and found that those regions are most often bound by AP1/IRF transcription factors. Forcing overexpression of cJun – a transcription factor that likely competes with both inhibitory and activating AP1 family members – reduced the expression of inhibitory receptors and increased IL2 production in CAR T cells exhausted by tonic signaling or excessive proliferative stress, and increased antitumor efficacy and persistence in multiple xenograft models. Interestingly, overexpression of cJun also enhanced CAR T cells’ ability to recognize antigen-low target cells.
Bispecific antibodies – protein fusions of an antigen-targeting domain and a T cell-targeting domain – are capable of redirecting a vast array of T cells to an antigen target on tumor cells and thus represent an opportunity for an off-the-shelf alternative to CAR T cells. Gregory Friberg reviewed Amgen’s experience with this therapeutic class, highlighting their experience with blinatumomab, the first product in this class to be FDA approved. Biologically, blinatumomab rapidly induced cytokine release, peaking in the first 24 hrs; caused a dip followed by an increase in peripheral T cells, peaking at 14-21 days; depleted peripheral B cells over the first week, mainly by apoptosis; and led to frequent complete elimination of acute lymphoblastic leukemia cells in the bone marrow over a 2-4 week period. Side effects included cytokine release syndrome, which is also observed with bispecifics for other targets and can be clinically managed once emergent, and neurological symptoms, which appeared to be CD19 specific and required immediate cessation of dosing. Retrospective analysis indicated that treatment of less bulky disease was more effective, especially in the minimal residual disease setting. Resistance was multifactorial, due to both host and tumor factors, and interestingly, loss of CD19 expression was uncommon, perhaps due to the on and off dosing schedule. Higher doses appear to be required for activity in tissues, compared to bone marrow, and the role of PD-1-mediated resistance is just beginning to be explored. Early clinical data with a bispecific antibody targeting CD33 also suggest targeting early disease may be potentially more effective, and early results with an anti-BCMA bispecific are encouraging.
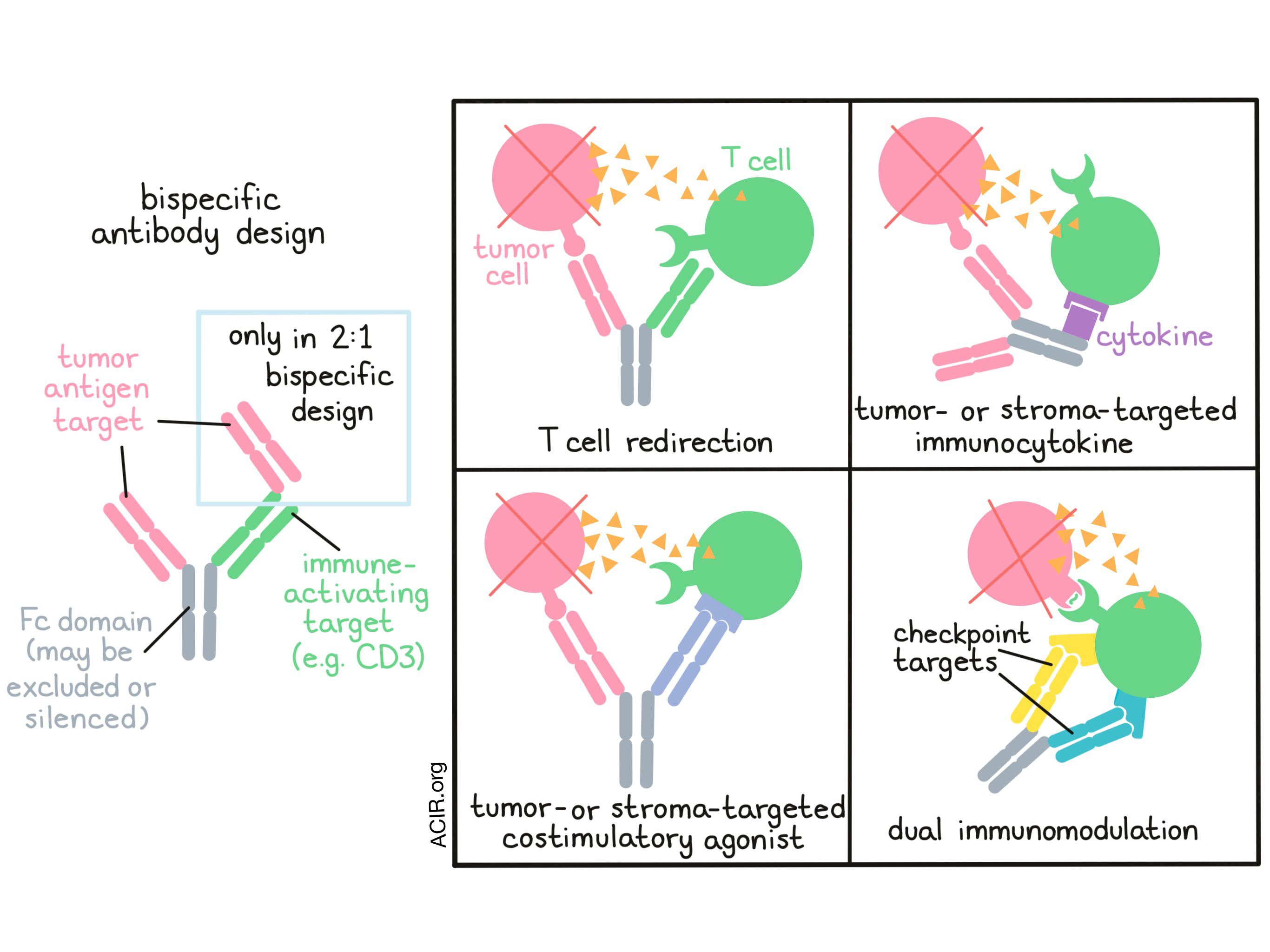
Providing an overview of bispecific antibodies and their current status in the clinic, Dominik Ruettinger of Roche emphasized the importance of design. According to Ruettinger, the most important aspect of a bispecific antibody is the chosen target antigen, followed by the antibody format and the properties of that format, the choice of Fc domain or exclusion of the Fc domain, and choice of the immune activating target (e.g. CD3, NKG2D, 4-1BB). Roche is currently developing bispecific antibodies with the potential to redirect T cells, modulate multiple immune checkpoints at once, or target cytokines or costimulatory agonists to the tumor. Ruettinger focused his talk on T cell bispecific (TCB) antibodies designed in a novel 2:1 format, with two antigen-targeting domains, one CD3 T cell engagement domain, and a silent Fc region to enhance the activity and to extend the half-life of the molecules. He presented data from a phase I dose escalation study of a CD20-TCB in patients with relapsed or refractory Non-Hodgkin’s lymphoma who were pretreated with obinutuzumab (anti-CD20 mAb) to mitigate toxicity associated with cytokine release syndrome. Ruettinger showed two examples of patients with diffuse large B-cell lymphoma who achieved deep partial responses. Similarly, from studies of cibisatamab (a carcinoembryonic antigen (CEA)-TCB) alone or in combination with atezolizumab (anti-PD-L1) against colorectal cancer, Ruettinger showed examples of dramatic inflammatory and antitumor responses. Not all patients in these trials derived clinical benefit, however, and Ruettinger suggested that antitumor responses to bispecific T cell engagers may largely depend on host immune fitness. To this end, Ruettinger showed that the new Roche prognostic (RoPro) score, which incorporates 26 variables reflecting lifestyle, host factors, and tumor stage, can be used to assess clinical trial populations. While patients may start with similar RoPro scores, treatment with a bispecific T cell engager induces early changes in RoPro scores that clearly separate responders and non-responders. Incorporating this scoring system could allow clinicians to quickly make decisions regarding whether to continue therapy or change course.
Expanding on the theme of T cell bispecific antibodies (TCB) in the 2:1 format, Pablo Umaña of Roche presented the results of a phase I clinical study with CD20-TCB (co-targeting CD20 and CD3). The molecule was safe with transient cytokine release symptoms and, as observed preclinically, pre-treatment with the anti-CD20 antibody obinutuzumab allowed immediate full dosing with the CD20-TCB. Durable CRs were observed in 29% of patients with aggressive lymphoma after as few as two cycles of treatment. Attempting to more powerfully stimulate antitumor immunity directly within tumors, Umaña described the design of a 4-1BB ligand-bearing, fibroblast-activating protein (FAP)-targeting antibody FAP-4-1BBL to target cancer-associated fibroblasts within the TME. Pre-clinically, the 4-1BBL stimulation enhanced activity in combination with other T cell-redirecting approaches; as a single agent, FAP-4-1BBL trafficked to the tumor tissue and showed encouraging signs of clinical activity in tumors with endogenous immunity.
Emerging Immunotherapy Approaches
In her discussion on “taming the beast” of the tumor immune microenvironment, Judith Varner snapped the whip at tumor-associated macrophages (TAMs), which tend to polarize towards an M2 state in the TME, mediate immune exclusion and suppression, and ultimately promote tumor growth. Varner suggested two options for targeting TAMs: inhibit the recruitment of bone marrow-derived macrophages to the tumor, or repolarize macrophages towards the pro-inflammatory M1 phenotype. Varner and her team identified PI3Kγ for its exclusive expression on myeloid cells and its role in myeloid cell recruitment during inflammation. Inhibiting PI3Kγ with IPI-549 depleted monocytes and granulocytes from the tumor and led to an influx of CD8+ T cells and activation of Th1 responses, but interestingly, left the macrophage population largely intact. While macrophages remained in the TME, the researchers noted that PI3Kγ inhibition activated NF-κB signaling, promoting a repolarization from an immune-suppressive to an immune-activating phenotype (enhanced MHC-II expression and cytokine production, particularly IL-12). A phase I clinical trial testing IPI-549 or IPI-549 + anti-PD-1 is currently underway, and some partial responses have already been observed, even in patients receiving monotherapy. Looking for additional ways to reduce suppressive macrophage in the TME, Varner and colleagues honed in on depleting tissue-resident macrophages, which derive from embryonic precursors that migrate to tissues and are resident for life. In early preclinical studies, the use of c-Kit inhibitors suppressed tissue-resident macrophage proliferation and controlled tumor growth in mice.
Given that most pediatric solid tumors have few mutations and low PD-L1 levels, single agent checkpoint blockade therapy is ineffective at reinvigorating the endogenous immune response. In the first part of his talk, Robbie Majzner explained that a more effective approach would be to create new immune responses with “synthetic” immunotherapies, specifically with B7-H3 CAR T cells. B7-H3 is highly and homogeneously expressed on pediatric solid tumors, including osteosarcoma, Ewing sarcoma, and medulloblastoma. In xenograft models, B7-H3 CAR T cells induced regressions of all three tumor types. In the second part of his talk, Majzner discussed combination targeting of CD47 (a “don’t eat me” signal) and GD2, which increases surface calreticulin (an “eat me” signal), to enhance the macrophage phagocytosis of tumor cells. The combination treatment synergized to induce long-term survival in mice with neuroblastoma or osteosarcoma xenografts.
Aurelien Marabelle argued that one way to enhance the efficacy of immunotherapy and to avoid on-target, off-tumor adverse events is with human intratumoral immunotherapy (HIT-IT). The safety of intratumoral delivery could allow for treatment with otherwise less safe combination therapies. Intratumoral administration can induce local priming, produce systemic antitumor responses, and significantly prolong survival compared with systemic administration. In situ immunization can be advantageous compared with other cancer vaccine approaches as it requires no antigen identification (making it more universal than personalized), does not necessitate biopsies, and can be manufactured off-the-shelf. The challenges of developing and testing HIT-IT include determining the most efficacious dose, rationally designing clinical trials, identifying relevant imaging assessment criteria, determining which tumor sites lead to the best responses when injected, and identifying predictive biomarkers of response. Marabelle stressed that the success of HIT-IT clinical trials depends on the ability to perform ancillary studies in parallel with clinical studies in order to answer some of these questions.
It is well known that cross-presentation of tumor antigens is crucial for the CD8+ T cell antitumor response, and high levels of the transcription factor Batf3 in DCs have been shown to be a major driver of cross-presentation. Joshua Brody discussed an early-phase clinical trial in which an intratumoral combination treatment (in situ vaccine) was used to enhance the antitumor T cell response: Flt3L recruited cross-presenting DC subsets to the tumor, local radiation enabled the release and uptake of tumor antigens by DCs, and poly-ICLC (a TLR3 agonist) activated the DCs. This combination treatment selectively activated myeloid cells within the TME and not in peripheral blood, thus making “cold” tumors “hot”. The treatment induced reproducible abscopal tumor regressions even in patients with advanced, high-tumor-burden disease due to recruitment of T cells to distant, untreated sites. Looking for factors behind differential response, Brody et al. found that exhausted CD8+ T cells were highly increased in the peripheral blood of non-responders after treatment, but were only mildly increased or even decreased in responders. In mice, the combination treatment induced PD-L1 expression and synergized with anti-PD-1 to increase survival, providing rationale for this quadruple combination treatment, which is currently in clinical trials.
Giorgio Trinchieri discussed the effect of the gut microbiome on inflammation, immunity, and response to cancer immunotherapy. Several recent studies have shown that certain types of bacteria confer favorable response to anti-PD-1 therapies, however, the particular bacteria responsible for this effect were completely discordant among the studies. Further analysis elucidated that these variable results were likely due to the differences in the regional diets of the cohorts. In addition to geography, cancer type and sequencing technology can affect microbe identification. Attempting to utilize machine learning to predict response revealed that while there was some predictive power within a cohort, that effect was lost when data from one cohort was used to attempt response prediction in another cohort. Therefore, there is a significant need to identify robust microbiome-related markers for predicting responders and to characterize shared mechanisms by which the microbiome enhances immunotherapy response. More discrete analysis of gene-level clustering showed some promise. In addition, identifying favorable microbiome compositions for fecal matter transfer and disecting how factors in patients’ diets (fiber, prebiotics, and probiotics) could induce or maintain a favorable microbiome can provide direct evidence of an effect. Finally, Trinchieri posed the question of how altering the microbiome could impact both efficacy of anti-PD-1 therapy and cachexia, a major contributor to cancer patient morbidity and mortality.
Elizabeth Jaffee gave the Presidential Address, in which she discussed the history and the current status of cancer immunotherapy. She noted that because many factors play a role in predicting and inducing individual response, there is a need for reliable predictive biomarkers, and cancer immunotherapy should be thought of as precision oncology treatment. Jaffee then discussed strategies for overcoming a “cold” immunological tumor microenvironment (TME), with a focus on the difficult-to-treat pancreatic cancer. Jaffee explained that the first step in converting a nonresponsive cancer to a responsive one is to induce T cell infiltration and activation, and this can be achieved with several approaches including vaccines, oncolytic viruses, radiation, and chemotherapy. The next step is to manipulate the immunosuppressive TME via agonist activation (e.g. targeting CD40, OX40, CD137) and/or checkpoint blockade (e.g. targeting PD-1/PD-L1, CTLA-4, TGFβ, IDO) in order to allow T cells to kill tumor cells. Jaffee summarized outcomes from a clinical trial combining an allogeneic, GM-CSF-expressing, irradiated whole tumor cell vaccine (GVAX) with anti-CTLA-4, as well as immunological outcomes and clinical correlates from another trial in which a similar vaccine was combined with anti-PD-1. She also emphasized the need for applying new technologies, such as multiplex immunohistochemistry and TCR sequencing, to identify new biomarkers for prediction of immunotherapy responses. Finally, Jaffee noted that inducing immunotherapeutic response in patients with advanced/metastatic cancers is time-sensitive due to short life expectancy, and that therapies should be rationally combined to induce tumor regression as soon as possible.
Antoni Ribas discussed the many forms of resistance to checkpoint blockade, and reviewed potential strategies for overcoming them. Although loss-of-function mutations leading to either dysfunction in the interferon signaling pathway or downregulation of MHC-I are rare, studying the underlying biology of these events has revealed useful approaches to overcoming resistance to checkpoint blockade. One example is forced expression of the MHC-I transactivator NLRC5, which bypassed the requirement for IFN signaling in MHC-I upregulation, and restored tumor sensitivity to adoptive cell therapy (ACT). Additionally, intratumoral administration of BO-112 (which stimulates TLR-3, RIG-I, and MDA-5) or the TLR-9 agonist SD101 induced responses in mice. In early clinical trials, the use of TLR-9 agonists in patients treated with anti-PD-1 have shown responses in both injected and uninjected lesions. Intratumoral delivery of the oncolytic virus T-VEC followed by pembrolizumab also induced high response rates in humans, independent of prior CD8+ T cell infiltration in advanced melanoma patients who had progressed on previous PD-1/PD-L1-based therapy. In tumors with β2M loss-of-function mutations that interfere with the antigen-presenting machinery, a kinetically engineered IL-2 receptor agonist NKTR-214 enhanced NK cell responses against tumors. Preclinically, in combination with ACT, NKTR-214 also improved antitumor activity by increasing systemic T cell expansion and tumor homing. NKTR-214 combined with anti-PD-1 therapy overcame immune resistance in mice bearing β2M-knockout MC38 tumors; 28 stage IV melanoma patients receiving the NKTR-214 + anti-PD-1 treatment as first line therapy showed a 50% ORR. To tackle resistance to checkpoint blockade mediated by T cell exclusion, PAK4 was identified as overexpressed in immune-excluded tumors and associated with deficits in T cells and dendritic cells. Genetic knockout of PAK4 reversed resistance to anti-PD-1 in B16 melanoma, dependent on CD8+ T cells, and researchers observed antitumor activity with use of the PAK4 inhibitor KPT-9274 in combination with checkpoint blockade.
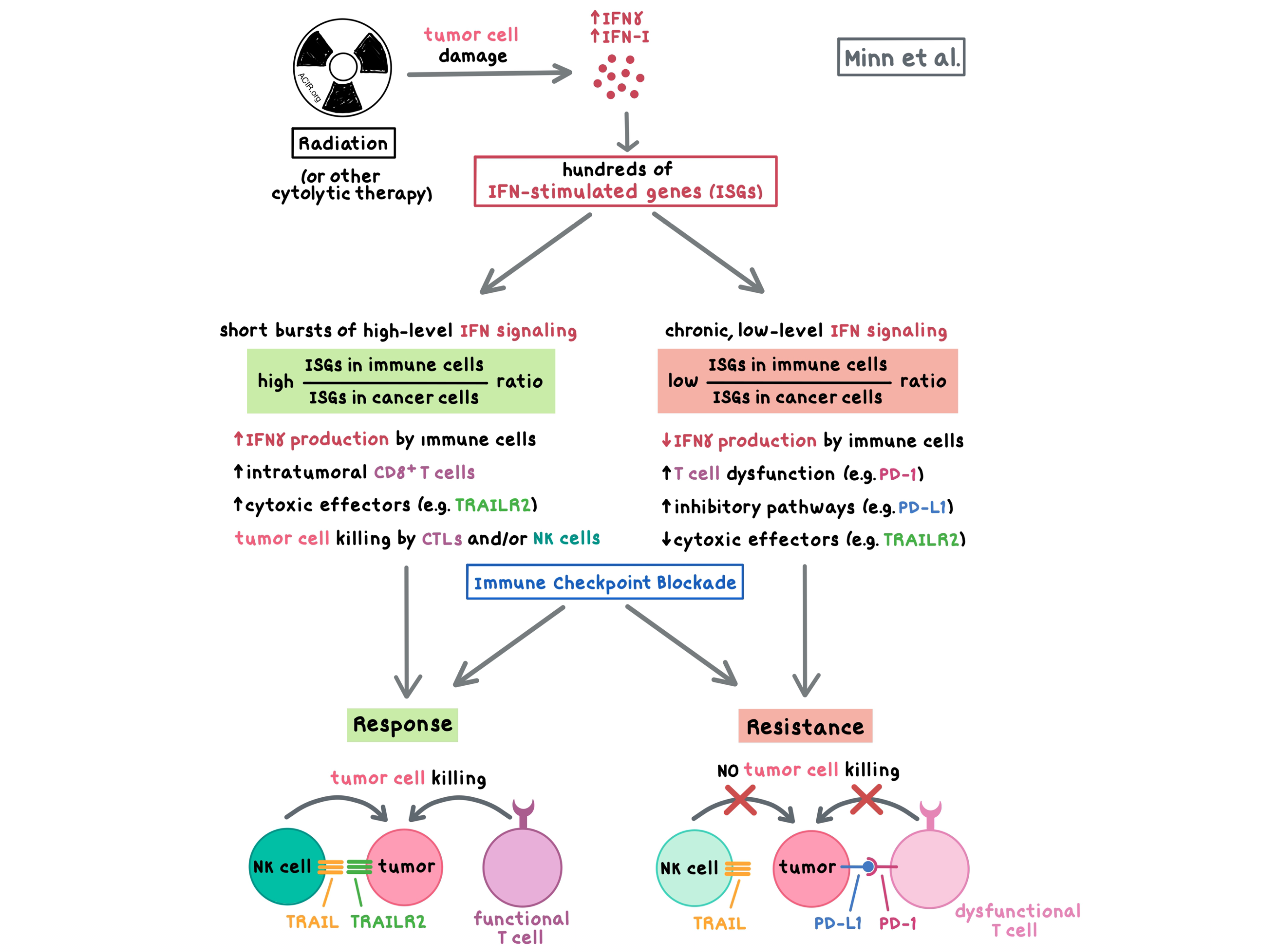
Andy Minn explained that, similar to the effects observed in acute vs. chronic infection, interferon (IFN) exerts dichotomous effects in cancer therapy, which depend on the magnitude and duration of IFN signaling: short bursts of high-level IFN signaling could lead to cancer cell death, while chronic, low-level signaling could induce resistance and T cell exhaustion. In a phase I clinical trial, patients with metastatic melanoma or NSCLC who had previously progressed on anti-PD-(L)1, treatment with anti-PD-1 and local radiation led to the regression of irradiated and unirradiated tumors in some patients, but the majority of patients did not respond. To understand the mechanism of resistance, Minn et al. turned to mouse models and found that although radiation and checkpoint blockade led to improved tumor control, tumors which escaped paradoxically had IFNGR-dependent higher expression levels of IFN-stimulated genes (ISGs) than the parental tumor. Chronic IFNγ signaling upregulated ISGs via epigenetic and transcriptomic changes. Importantly, ISGs in cancer cells led to an increase in inhibitory pathways, a decrease in cytotoxic effectors, increased T cell dysfunction, and decreased immune cell IFNγ signaling, ultimately resulting in relapse and resistance. Inhibition of IFNγ signaling in cancer cells increased intratumoral CD8+ T cells, enhanced IFN signaling in immune cells, and mediated tumor cell killing by cytotoxic T lymphocytes (in tumors with adequate neoantigens and MHC-I expression) and NK cells (which could be active in neoantigen- and MHC-I-poor tumors), thus improving response to immune checkpoint blockade in several mouse models. The ratio of ISGs in immune cells to ISGs in cancer cells predicted response independent of tumor mutation burden. In NSCLC patients treated with anti-PD-1 and anti-CTLA-4, certain mutations in the IFN pathway predicted ICB response and resulted in prolonged progression-free survival.
Given that high CD8+ T cell infiltration predicts longer progression-free survival in patients with renal cell carcinoma (RCC), Caroline Jansen (from Haydn Kissick’s lab) examined the CD8+ T cell subsets in tumor tissue collected from 68 RCC patients undergoing surgery and found that the CD8+ TILs consisted of CD28+TCF1+Gzmb- stem-like cells and CD28-TCF1-Gzmb+ effector-like cells; both were important for the antitumor response. The two subsets were clonally related, and Jansen et al. found that the stem-like TILs were highly proliferative and were able to not only self-renew but also to differentiate into the effector-like TILs, thus maintaining both populations within the tumor. Intratumoral antigen-presenting niches containing a high density of DCs and stem-like CD8+ TILs were necessary for the maintenance of the CD8+ T cell response.
Although lymph nodes (LNs) are sites of antigen presentation and education of lymphocytes, LN metastasis correlates with disease progression and poor outcome, and not with increased antitumor response. To understand how LN metastasis leads to distant metastases, Nathan Reticker-Flynn (from Edgar Engleman’s lab) implanted B16-F0 melanoma into mice, isolated clones that spread into the LNs, and performed 9 rounds of re-implantation to generate 300 cell lines with an enhanced capacity to metastasize to the LN. Reticker-Flynn et al. showed that the LN cell lines were enriched in an interferon response gene expression program and that this signature was necessary to colonize the LN, with the LN metastases forming adjacent to the active germinal centers. The LN cell lines interacted with lymphocytes within the LN, induced Tregs, and promoted metastatic seeding to distant tissues by disseminating tolerance. Depletion of Tregs greatly reduced tumor growth and eliminated distant organ metastasis formation.
Challenging the current understanding of the mechanism of action of PD-1/PD-L1 blockade, Yuting Liu (from David Rimm’s lab) presented evidence that PD-L1 on host macrophages in both the tumor microenvironment and stroma drive the efficacy of PD-L1 blockade, rather than PD-L1 on tumor cells. After citing prior research showing that host PD-L1 expression is essential for the efficacy of PD-L1 blockade, Liu showed images taken using confocal microscopy, which demonstrated qualitatively that what might typically be measured as tumor PD-L1 could actually be PD-L1 expressed on macrophages hidden behind a “tumor mask”. Quantitative analysis using inForm showed that 40% of PD-L1 in the tumor was expressed on macrophages, and 80% of PD-L1 detected in the stroma was expressed on macrophages. To test the predictive power of host PD-L1 as a biomarker, Liu and colleagues analyzed data from patients in a 67-patient cohort treated with anti-PD-1 or anti-PD-L1 monotherapy. While high PD-L1 on macrophages was associated with high PD-L1 expression in the tumor, only PD-L1 expression on macrophages correlated with better overall survival in these patients. The predictive value was validated using several methods of analysis, and similar results were observed using Nanostring Digital Spatial Profiling in an unrelated cohort of melanoma patients. In a proposed mechanism of action, Liu suggested that PD-1 on activated T cells in the TME may bind PD-L1 on macrophages, suppressing macrophage production of T cell-attracting chemokines and cytokines. PD-1/PD-L1 blockade overcomes the suppression, inducing macrophages to increase production of chemoattractants, drawing T cells to the tumor site.
Cornelis Melief discussed the results of several clinical trials involving ISA101 – a therapeutic synthetic long peptide HPV16 E6 and E7 vaccine. The vaccine had previously been shown to induce durable complete remissions and partial responses in more than 50% of patients with premalignant vulval intraepithelial neoplasia. However, the vaccine was less effective in advanced cancers due to the immunosuppressive TME. In a pilot study of 18 patients with recurrent/metastatic HPV16+ cervical cancer, the combination of carboplatin/paclitaxel chemotherapy and a single dose of ISA101 decreased the levels of circulating immunosuppressive myeloid, but not lymphoid, cells and induced a sustained HPV16-specific T cell response. In a single-arm phase I-II clinical trial, 77 patients with advanced, metastatic, or recurrent HPV16+ cervical cancer were treated with 6 cycles of carboplatin/paclitaxel and 3 doses of ISA101. The combination therapy led to a significant reduction in tumor size in many treated patients. A pivotal phase III trial for assessing the efficacy of triple therapy combining chemotherapy, vaccination, and checkpoint blockade as first-line therapy for metastatic HPV16+ cervical cancer is being planned.
David Andrews discussed the results of a phase Ib clinical trial in which 32 patients with newly diagnosed glioblastoma (GBM) were treated with a personalized autologous whole tumor cell vaccine within 24 hours of craniotomy. The vaccine consisted of abdominally implanted diffusion chambers encapsulating irradiated GBM tumor cells together with an antisense oligodeoxynucleotide molecule against insulin-like growth factor type 1 receptor. During the dose escalation phase, the highest dose conferred the strongest induction of pro-inflammatory serum cytokines and the most promising indication of clinical benefit; the remaining patients were treated with the highest dose. Several grade 3 adverse events were observed, though none were attributed to the vaccine. Best responses showed no local recurrence at resection site after total resection, and regression of the tumor to non-measurable disease after a subtotal resection. Patients with good preoperative T cell function (as assessed by IFNγ production) experienced longer overall survival. The vaccine led to improved overall and progression-free survival compared with institutional or published historical standard-of-care treatment, particularly for patients with MGMT promoter methylation. A phase II dose escalation study is planned.
Lakshmi Srinivasan of Neon Therapeutics discussed the results from a clinical trial of the personalized neoantigen vaccine NEO-PV-01 combined with nivolumab in patients with metastatic melanoma who were naive to PD-1/PD-L1 treatment. Combination therapy induced neoepitope-specific CD4+ and CD8+ T cell responses to an average of 55% of the peptides included in the vaccine. Each patient demonstrated responses to multiple peptides and all patients demonstrated ex vivo detectable responses. Vaccine-induced T cell responses were typically mutation-specific. In one case study, the T cells were shown to be polyfunctional and cytolytic, and exhibited effector memory and memory phenotypes, likely contributing to the durable vaccine-induced immune responses that were observed. In a separate case study neoantigen-induced T cells were demonstrated to track to the tumor. Further, epitope spreading – new T cell responses to neoantigens not included in the vaccine – was observed in 8/8 patients who achieved a durable clinical benefit (DCB). A number of additional factors were found to be associated with DCB, including a high number of mutations with a high epitope quality score; strong pretreatment gene signatures for MHC-II, B cells, and TCF7+CD8+ T cells; and a pathologic response following vaccination.
Julius Strauss discussed the results of an ongoing clinical trial testing M7824 (a.k.a. bintrafusp alfa), a bifunctional fusion protein targeting both TGFβ and PD-L1, in 43 checkpoint blockade-naive patients with advanced HPV-associated cancers. Immune-related adverse events were largely similar to those observed in PD-1/PD-L1 blockade, though some adverse events consistent with TGFβ blockade (low grade squamous cell carcinomas of the skin and low-grade mucosal bleeding) were also observed and the overall safety profile was considered manageable. Altogether, 2 patients achieved complete responses, 10 achieved partial responses, and 3 more achieved delayed partial responses after progressive disease. Among all patients, the objective response rate (ORR) was 34.9%. Among 36 patients with confirmed HPV+ status, the ORR was 38.9%. Most responses were durable, with 11 of the 15 responses ongoing (4-22+ months) at the time of data cutoff. Interestingly, responses occurred regardless of PD-L1 expression status.
Drawing attention to immune-modulating molecules beyond checkpoint blockades, Roberta Zappasodi discussed the use of immune-stimulating antibodies engaging the TNF-receptor family member GITR to enhance T cell proliferation and effector functions and knock down Tregs. Based on promising preclinical data, Zappasodi and colleagues launched a phase I trial of a humanized agonistic anti-GITR antibody in 37 patients with advanced cancers. While treatment frequently reduced circulating and intratumoral Tregs (likely via hyperactivation and destabilization leading to Foxp3 loss and cell death), no significant clinical responses were observed. Moving back into mice, the researchers found that treating B16F10 melanoma-bearing mice with anti-GITR on day 4 was curative, while treating mice on day 7 was refractory, despite similar reduction in Tregs and an increase in the CD8+:Treg ratio in both groups. Gene expression analysis indicated that CD8+ T cells in the refractory group expressed high levels of exhaustion markers and low levels of memory and functional markers, suggesting a rationale for combination with PD-1 checkpoint blockade. Administration of combination anti-GITR + anti-PD-1 at the 7 day time point (refractory to either monotherapy) depleted Tregs and reinvigorated previously exhausted CD8+ T cells, yielding a 50% complete response rate in mice and warranting the start of a clinical trial, currently underway, of this combination strategy.
Olivera Finn discussed the results of a phase II clinical trial that tested the efficacy of a preventative MUC1 vaccine in 110 patients with newly diagnosed, endoscopically resected, advanced adenoma (a precursor to colon cancer). MUC1 is a shared, non-viral, tumor-associated antigen that becomes abnormally glycosylated and overexpressed in adenomas and in colon carcinomas. Finn et al. hypothesized that the MUC1 vaccine, which is weakly immunogenic in late stage cancers, would be more immunogenic in premalignant disease, a setting in which the immune system is less suppressed. Patients were randomized to receive either placebo or MUC1 + poly-ICLC at weeks 0, 2, and 10, and a booster at week 52. The vaccine was well tolerated. Patients treated with the vaccine were much more likely than those treated with placebo to develop an anti-MUC1 IgG immune response at week 12, and all patients that responded at week 12 also responded at week 55 after the booster, indicating vaccine-induced immunological memory. These results were consistent with a previously conducted small pilot study, which had also indicated strong T cell immune memory. Non-responders exhibited increased pre-vaccination levels of circulating MDSCs, suggesting the presence of immunosuppression even in premalignant lesions and the need to vaccinate as early as possible. Further analyses will show whether the vaccine can reduce polyp recurrence and whether vaccination affects the immune microenvironment of recurrent adenomas.
by Anna Scherer, Lauren Hitchings, and Ed Fritsch



