
Last week, the ACIR team attended the AACR Annual Meeting 2023 in Orlando, Florida. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Personalized cancer vaccines
Jeffrey S. Weber
Vinod P. Balachandran
Catherine J. Wu
Tumor immune microenvironment and beyond
Gabriel A. Rabinovich
Ignacio Melero
Karin E. de Visser
Florencia McAllister
Greg M. Delgoffe
T cells - Biology and Therapy
Katelyn T. Byrne
Eric Tran
Judith Agudo
Amanda W. Lund
Attaya Suvannasankha
Checkpoint blockade and combination therapies
Valsamo K. Anagnostou
Eduardo Castañón
Elizabeth M. Jaffee
Myeloid cells and DC therapy
Michele De Palma
Judith A. Varner
Personalized cancer vaccines
A personalized cancer vaccine, mRNA-4157, combined with pembrolizumab versus pembrolizumab in patients with resected high-risk melanoma: Efficacy and safety results from the randomized, open-label Phase 2 mRNA-4157-P201/Keynote-942 trial - Jeffrey S. Weber - NYU Langone Medical Center, New York, NY
The emergence of neoantigens as truly tumor-specific, immunogenic, and safe vaccine targets over the past decade has breathed new life into the field of cancer vaccines, particularly after disappointing large trials utilizing tumor-associated antigens. Neoantigen-based vaccines elicited strong, lasting T cell responses and even epitope spread, a key marker of on-target vaccine activity. Recognizing that checkpoint blockade antibodies have now become the standard of care for multiple diseases (such as high-risk melanoma), but still require improvement in both response rates and durability, Jeffrey Weber described the very encouraging results of a randomized phase 2 clinical trial comparing checkpoint therapy alone (pembrolizumab) to pembrolizumab with a personal neoantigen mRNA-based cancer vaccine (mRNA-4157) in patients with high-risk, resectable melanoma. Using nucleic acid sequencing of patient tumor and normal samples to identify patient-specific mutations, advanced algorithms to select promising neoantigen targets, and an integrated manufacturing process to prepare mRNA encoding (mostly) 34 personal neoantigens targets for each patient, 157 patients were randomized (2:1) to receive the vaccine plus checkpoint combination or checkpoint therapy alone every three weeks. The combination therapy was safe, with an increase in mild AEs compared to pembrolizumab alone, characterized as a “typical COVID vaccine”-type response. Remarkably, combination therapy led to a significant (one-sided p=0.026) increase in relapse-free survival (RFS) with a hazard ratio of 0.56 and a 44% reduction in risk of recurrence over time. The RFS curves began separating at about 9 months, and the 18-month RFS was 79% compared to 62% in the control arm. The improvement seen in the combination arm was independent of the PD-L1 status of the patients, with some indication that the effect was larger in PD-L1-low patients. Separate data presented by Ryan Sullivan showed that it was also independent of tumor mutation burden. Based on these results, plans are underway to initiate a phase 3 study in 2023, and to expand into other immunotherapy-responsive histologies, such as NSCLC.
Individualized mRNA neoantigen vaccines for pancreatic cancer - Vinod P. Balachandran - Memorial Sloan Kettering Cancer Center, New York, NY
Vinod Balachandran studies pancreatic ductal adenocarcinoma (PDAC), which is the quintessential example of an immunologically cold tumor, with a low mutation burden, minimal T cell infiltration, and resistance to immunotherapy. However, around 9% of PDAC tumors are hot, and patients with such tumors have better survival. With the objective of identifying neoantigens that could most effectively prime de novo responses, amplify pre-existing responses, or both, Balachandran and colleagues developed a model for evaluating the quality of neoantigens by estimating “selfness” and “non-selfness” based on predicted MHC binding, potential TCR reactivity, and similarity to known viral antigenic peptides. Interestingly, the quality of neoantigens was better at predicting patient survival than the quantity of neoantigens, and T cells specific for high quality neoantigens could be identified in the blood of a number of long-term survivors of pancreatic cancer up to a decade later. The team also found that high quality neoantigens were likely to be edited in recurring tumors of long term survivors, and that the spontaneous development of high quality neoantigens could lead to increased neoantigen-specific T cells that may delay tumor recurrence. Given that nearly all PDACs harbor pools of neoantigens with varying predicted immunogenicity, Balachandran and colleagues wondered whether RNA vaccines could be used to spark de novo responses. In a collaborative study, they developed individualized mRNA vaccines for pancreatic cancer. In a phase I trial, patients underwent surgery, and were later treated with atezolizumab at week 6, followed by 8 consecutive doses of a personalized cancer vaccine starting at week 9, and then standard-of-care chemotherapy starting at week 21, with a final late booster at week 46. While the trial was disrupted by the COVID-19 pandemic, 16 out of 19 patients who received atezolizumab went on to receive their vaccines, supporting the feasibility of the generation and administration of a custom vaccine after surgery. Primary ELISpot analysis and confirmatory TCR sequencing CloneTrack tests were used to monitor for vaccine-induced T cell responses, and together revealed robust post-vaccination responses that could be maintained even after chemotherapy. Clustering clones based on vaccine-induced clonal trajectories in peripheral blood showed that the polyclonal T cell pool expanded by the mRNA vaccines included clones that were undetectable prior to vaccination, and was distinct from the pool expanded by anti-PD-L1 – an effect that was particularly apparent after vaccine boosters, which further expanded the vaccine-induced, but not PD-L1-induced clones. Further, the researchers saw that vaccine-expanded clones were not found in tumors harvested prior to vaccination, suggesting that vaccines might be priming de novo responses. Further analysis revealed that many of these T cell clones expanding in the peripheral blood were, in fact, neoantigen-specific, even in patients with less clonal expansion overall. Patients who showed both primary ELISpot activity and confirmatory clonal expansion were classified as mRNA neoantigen vaccine responders. Overall, mRNA vaccines induced T cell responses in 50% of PDAC patients, many of which were substantial and durable. At a median follow-up of 18 months, responders had not yet reached a median recurrence-free survival, while non-responders had a median recurrence-free survival of 13.4 months. A similar correlation with recurrence was not observed for atezolizumab treatment, differences in standard prognostic variables (lymph node status or resection margins), chemotherapy cycles, or T cell density in primary tumors. Considering that non-responders may be less fit for mRNA vaccination, the researchers compared patient responses to the COVID-19 vaccine, and found that cancer vaccine responders and non-responders mounted similar responses to the COVID-19 vaccine, ruling out baseline immune fitness as a contributing variable. They did, however, find that responders had more clonal mutations and higher quality neoantigens. In a clinical vignette, Balachandran described a patient who appeared to have evidence of metastasis, along with a suspicious liver lesion, which ended up being a lymphoid aggregate of vaccine-expanded T cell clones that disappeared with later imaging. This result may represent vaccine-induced immune control of a micrometastasis. Moving forward, Balachandran and colleagues will be conducting a phase 2 clinical trial of this vaccine strategy, and are considering ways to scale this treatment to other checkpoint blockade-resistant cancers.
T-cell specificity, phenotype, and dynamics: impact on cancer vaccines - Catherine J. Wu - Dana-Farber Cancer Institute, Boston, MA
Catherine Wu, a newly appointed Fellow of AACR Academy 2023, began her talk by discussing how cancer vaccination has the potential to address a number of challenges to antitumor immunity, from tumor heterogeneity and evolution, to immune unresponsiveness, as cancer vaccines can be used to both access pre-existing tumor immunity and induce new responses. One important obstacle in cancer vaccine development is the identification of tumor-specific TCRs, which are relatively rare in the native landscape and are mixed in with bystander T cells in the TME. For Wu and team, identifying relevant tumor-specific T cells has involved integrating considerations of the TCR sequence and the T cell phenotype based on advances in single-cell sequencing technology and the enhanced identification of potential target antigens via mass spectrometry and other techniques. This linkage was enabled by utilizing a combinatorial dye labeling system that allows for high throughput screening of a large number of TCR candidates for specificity to tumors and target antigens. Evaluating the TIL landscape in data from four patients with melanoma, Wu and colleagues used clustering analysis to map the distinct CD8+ T cell phenotypes and focused on the two largest populations: exhausted effector cells and non-exhausted memory cells. Reconstructing TCRs from the dominant clonotypes in each of these groups, the researchers observed much higher tumor specificity in the exhausted T cells than in the non-exhausted memory cells, which were more enriched for viral specificities. CD4+ T cells could also be split into distinct phenotypes, categorized as exhausted, non-exhausted memory, or Tregs. Interestingly, the highest tumor specificity was observed in Tregs. Each of the four evaluated patients showed very different profiles of antigen specificity, demonstrating a range of specificities for both melanoma-associated antigens (MAAs), neoantigens, and viral antigens, with neoantigen-specific TCRs showing much higher avidity compared to the MAA-specific TCRs. Beyond melanoma, the researchers observed similar clustering results in data from renal cell carcinoma (RCC) and head and neck squamous cell carcinoma (HNSCC). Evaluating tumor-reactive T cells from a different angle, Wu and colleagues investigated the spatial cellular relationships of cells in the TME using technologies built on Slide-seq, which allows for analysis of RNA, DNA, and TCRs in tissue sections. Using this system, the researchers were able to identify TCRs that were depleted or enriched within tumors compared to the tumor margins. Combining Slide-seq with TCRseq in human melanomas, the researchers could analyze multiple regions of a sample and identify tertiary lymphoid structures. One interesting sample consisted of two lobes, and showed evidence of a TCR that was restricted to one lobe, demonstrating a spatial bias. Looking at the relationship to its environment, this was positively associated with the chemoattractant CXCL10, and negatively associated with MGST1, which has previously been associated with response to immunotherapy. Overall, looking at the TIL landscape of melanoma, it became clear to Wu that the T cells with specificities that would be most beneficial in immunotherapy are, unfortunately, exhausted. Investigating ways to revive or replace them, Wu turned to the identification of personalized neoantigens and the generation of personalized cancer vaccines. In clinical works published in 2017 by two separate groups, peptide vaccines and RNA vaccines targeting personalized neoantigens each demonstrated safety, feasibility, immunogenicity, and promising efficacy when used in combination with checkpoint blockades. Despite the different delivery mechanisms, similar results were observed between the two studies, and some complete responses from those studies have continued to endure. Long-term follow-up of patients’ immune profiles 3-4 years after treatment showed persisting memory T cell responses to vaccine neoantigens, along with T cell diversification and evidence of epitope spreading. While acknowledging areas that need improvement, including time and cost at scale, Wu remains optimistic that personalized antigen-targeting vaccines could provide solutions to a number of challenges to immunotherapy, especially when used in rational combinations, possibly even approaching prevention.
Tumor immune microenvironment and beyond
Reprogramming the tumor-immune microenvironment by targeting galectin-glycan interactions - Gabriel A. Rabinovich - Instituto de Biología y Medicina Experimental, Ciudad de Buenos Aires, Argentina
In "a sweet adventure from south of the equator" Gabriel Rabinovich presented his decades-long research on Galectin 1 (Gal-1). Gal-1 is upregulated in most human tumors and correlates with poor prognosis. It is expressed and secreted by cancer cells, fibroblasts, and some immune cells, and has two binding sites for glycan N-acetyl-lactosamine (LacNac), present in complex N-glycans and core 1/2 O-glycans. Gal-1 acts as a glycocheckpoint that promotes tumor immune escape and angiogenesis. Gal-1-gylcan interactions reprogram the immune landscape in cancer by inducing selective death of Th1, Th17, and CD8+ T cells, while sparing Th2 and Treg cells carrying α(2,6)-sialylated cell surface glycoproteins, promoting tolerogenic dendritic cells via IL-27- and IL-10-driven circuits and M2 macrophage polarization, and activating immunosuppressive and pro-angiogenic programs in MDSCs. Gal-1 also links immune suppression and angiogenesis in the tumor microenvironment. Under hypoxic conditions, cancer cells secrete Gal-1, which can bind to VEGFR2 on endothelial cells, mimic VEGF, and promote vascularization. Increases in circulating Gal-1 levels in bevacizumab (anti-VEGF mAb)-treated patients with melanoma was associated with higher risk of recurrence and death in the AVAST-M phase 3 trial, and plasma samples from non-responding patients could reprogram endothelial cells through the Gal-1-VEGFR2 pathway. In summary, a network of Gal-1 and other galectins control the immune and vascular landscape in the tumor microenvironment and influence several hallmarks of cancer. Galectin-driven circuits in cancer can be targeted by small molecule carbohydrate inhibitors, natural polysaccharides and their derivatives, peptides and peptidomimetics, neutralizing monoclonal antibodies, and truncated galectins. Many of these approaches are currently being investigated in various phases of clinical trials. A Gal-1-specific neutralizing mAb developed by Rabinovich inhibited tumor growth and angiogenesis and counteracted resistance to anti-VEGF treatment in mouse models. It also increased the influx of immune cells (particularly, CD8+ T cells) into the tumor and induced reprogramming of MDSCs.
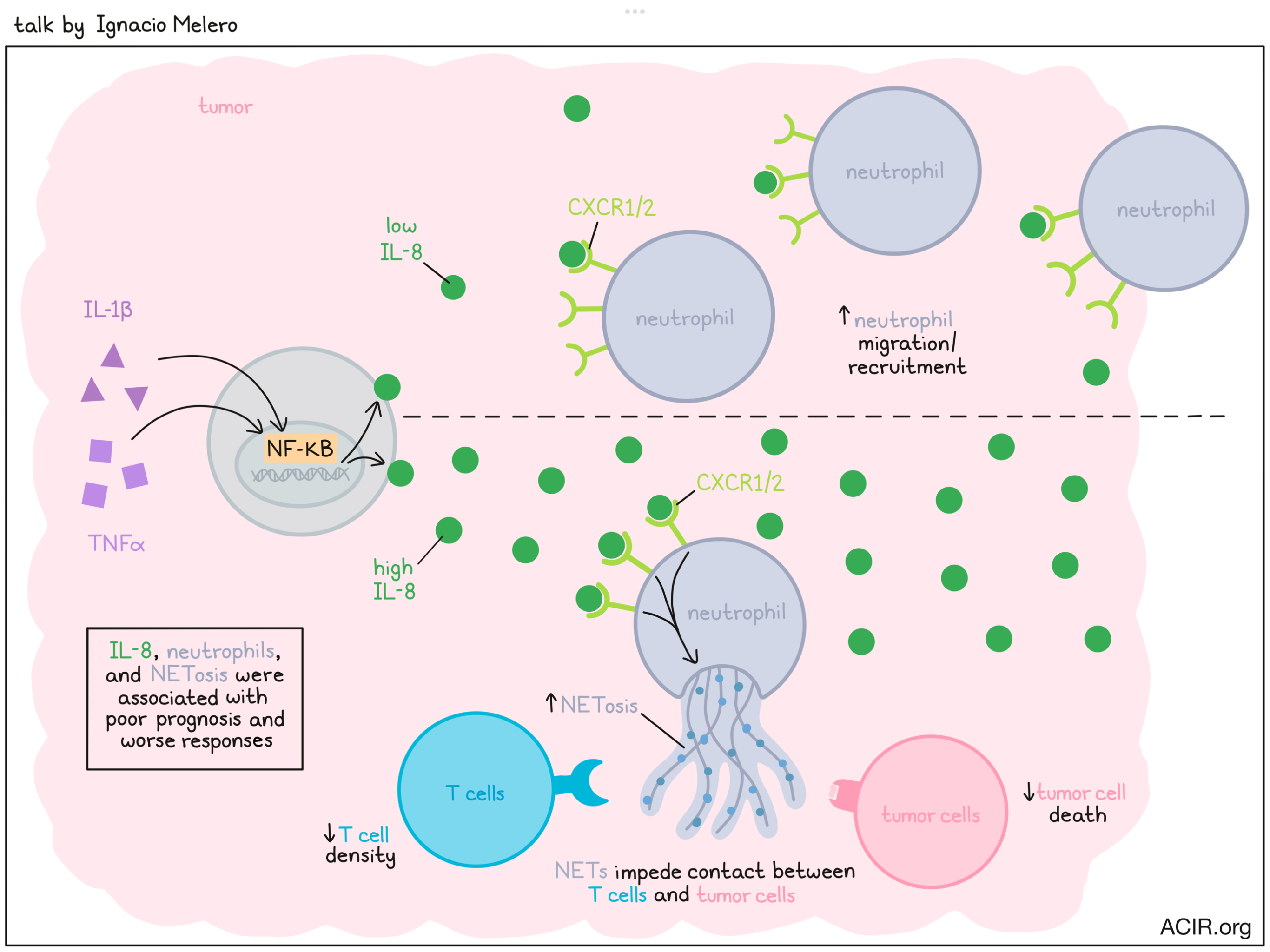
Interleukin-8, neutrophils and NETs in a druggable collusion against cancer immunotherapy - Ignacio Melero - Clínica Universidad de Navarra, Pamplona, Spain
The role of the immune system in cancer is complex and often contradictory, as some immune cells work to fight the cancer, while others work to protect it. This, as Ignacio Melero put it, requires immunotherapists to be both arsonists and firefighters at the same time. Focusing on suppressing undesirable immune responses, Melero honed in on neutrophils, which can play multiple roles, but are typically considered pro-tumor and associated with poor prognosis. Neutrophils are capable of degranulation, phagocytosis/endocytosis, production of reactive oxygen species, and a process called NETosis, in which they release DNA coated with proteins and polypeptides, forming a sticky net-like structure that can trap and kill microbes. In the setting of cancer, neutrophil extracellular traps (NETs) have a known role in supporting metastasis and inhibition of immune responses. Melero and colleagues found that these NETs likely help malignant cells to survive by impeding physical T cell contact with target cells, protecting cancers. Looking at IL-8, which is known to attract neutrophils, the researchers found that in data from multiple clinical trials, high IL-8 expression in patients with cancer correlated with worse responses to immune checkpoint blockade, and could even serve as a biomarker for responses, independent of PD-L1 and tumor mutation burden. IL-8 also correlated positively with the presence of neutrophils, NETosis, and monocytes in tumors, and negatively with T cells and IFNγ. Looking more closely at concentrations of IL-8, the researchers showed that low levels of IL-8 induced neutrophil migration and recruitment, while high levels of IL-8 were responsible for inducing NETosis. Given that IL-8 interacts with CXCR1 and CXCR2 receptors, Melero and others showed that blockade of CXCR1/2 or of signaling molecules downstream of CXCR1/2 could reduce NETosis in tumor organoids, mice bearing murine tumors, and humanized mice bearing human tumors. Importantly, this blockade also improved CD8+ T cell- and NK cell-mediated killing of tumors. In line with this, NET expression across different tumors correlated positively with IL-8 production and negatively with CD8+ T cell density; similar correlation could be made by measuring NETs in serum. Next investigating what drives increased IL-8 production in tumors, Melero turned towards NF-κB, which is induced by TNFɑ and IL-1β. Looking at these cytokines (IL-8, TNFɑ, and IL-1β), expected correlations were observed in serum, with similar effects in mice (using CXCL1 and CXCL2 as IL-8 orthologs). Further, blocking or injecting TNFɑ and IL-1β could effectively block or promote IL-8, respectively. Clinical relevancy of this was demonstrated in samples from patients treated with an anti-TNFɑ antibody, which showed reduced IL-8 serum levels 24 hours after treatment. Clinical trials investigating the blockade of CXCR1/2 or IL-8 are currently ongoing, and while high doses have been required to completely neutralize IL-8, treatment thus far has been safe, even in combination with nivolumab plus ipilimumab. Further, reductions in neutrophils in treated tumors have been observed.
Crosstalk between eosinophils and adaptive immune cells: A new avenue for immune checkpoint blockade in breast cancer - Karin E. de Visser - Netherlands Cancer Institute, Amsterdam, Netherlands
Immune checkpoint blockade (ICB) has proven effective against solid tumors; however, the response rate in patients with advanced breast cancer is disappointingly low, with only 5-10% of patients responding. Although patients with PD-L1-positive triple-negative breast cancer (TNBC) show an improved response to ICB plus chemotherapy regimens, the majority of patients still do not respond to the ICB. While most studies on ICB response focus on T cells, the crosstalk between innate and adaptive immune cells is critical for an effective immune response. Karin de Visser and colleagues performed unbiased profiling of immune cell dynamics in longitudinal samples obtained from the blood and metastatic lesions of ICB-treated patients with TNBC on the TONIC clinical trial. Together with accompanying mouse studies, this allowed them to investigate immune parameters associated with response. A systemic and intratumoral increase in eosinophils was observed in responding patients, but not non-responding patients, upon ICB treatment. A more than 2-fold increase in systemic eosinophil levels correlated with better overall survival in the TONIC clinical trial. Genetically engineered KEP (K14-cre;Cdh1f/f;Trp53f/f) mice developed spontaneous invasive breast cancer that was not responsive to ICB alone, but was responsive to the combination of ICB and cisplatin in a T cell-dependent manner, similar to what is observed in patients with TNBC. In this mouse model, the ICB and cisplatin combination led to both systemic and intratumoral increases in eosinophils. Depletion of eosinophils with an anti-SiglecF antibody abolished the therapeutic benefit of ICB plus cisplatin. In another KEP-based multi-organ metastatic disease model, ICB plus cisplatin induced a durable response in an eosinophil-dependent manner. To understand how the therapy mobilizes eosinophils, de Visser and her team analyzed plasma cytokines, and found that IL-5 levels were upregulated in ICB-treated mice and that IL-5 neutralization abrogated the therapeutic benefit of ICB. Since CD4+ T cells are the main producers of IL-5, the researchers FACS-sorted CD4+ T from ICB-treated patients with TNBC and found upregulation of IL-5 mRNA in these cells. Depletion of CD4+ T cells led to the loss of the eosinophil signature in mouse models, suggesting that ICB treatment induces IL-5 in CD4+ T cells, which in turn increases eosinophil production in the bone marrow, and systemic eosinophil accumulation and activation. To determine the factors responsible for the recruitment of eosinophils into the tumors, the researchers analyzed cytokines in the plasma of cisplatin plus ICB-treated patients and found that the alarmin IL-33, a known chemoattractant of eosinophils, was only upregulated upon cisplatin treatment. IL-33 neutralization abolished the intratumoral accumulation of eosinophils and reduced tumor control in mice. Similarly, IL-33 levels correlated with an eosinophil gene signature in tumor samples of ICB-responding, but not non-responding patients. Systemic treatment of mice with recombinant IL-33, to mimic the effect of cisplatin, increased intratumoral eosinophils, but required the combination with ICB to activate CD8+ T cells. In an alternative strategy, Treg depletion led to intratumoral eosinophil accumulation, and again the combination with ICB treatment was required to improve survival in the metastatic KEP-based tumor model. To summarize, ICB plus cisplatin therapy induces CD4+ T cells to produce IL-5, which increases eosinophil production in the bone marrow, leading to systemic accumulation and activation of eosinophils that are recruited by IL-33-producing tumors and improve ICB response in breast cancer.
Mechanisms employed by the gut and tumor microbiome to mediate responses to immunotherapy - Florencia McAllister - UT MD Anderson Cancer Center, Houston, TX
Microbes of many types, but particularly bacterial species, are abundant at all body barriers with mucosal tissues or where body barriers may be disrupted by pathogenesis, and so may have important effects across a variety of cancer types. Studies on the impact of gut microbiota on antitumor immune responses have led the way in this field. Florencia McAllister began by briefly reviewing published studies by her group and others, with an emphasis on the importance of controls (cohorts across geographies, collection methods, etc.), application of multiple existing and developing tools to increase confidence, and searching for function and mechanistic information that may allow more targeted approaches in the future. Focusing on tumor-resident microbes (TRMs), important new tools include spatial assessments of microbes with respect to tumor and immune cells, and quantitative and qualitative species-identifying new nucleic acid approaches. Assessing the functionality of TRMs by recovery of viable organisms and in vivo modulation of the composition are also critical, and McAllister provided some seminal examples of the interactions between microbes and drug therapies, the effects of immune-activating or -inhibiting microbes on survival in pancreatic cancer, and the relationship between microbes and primary and metastatic tumors. Turning to her newer work, McAllister described developing a tool to quantitate a microbial/host DNA ratio to broadly measure overall intratumoral microbial tumor burden (ITMB). This tool was applied to TCGA data across 4 tumor types (gastric, colorectal, breast, and uterine), and interestingly, a high ITMB was associated with multiple immune pathways, but only in the gut-related gastric and colorectal tumors. Furthermore, high ITMB was associated with increases in activated mast cells and decreases in resting mast cells. These results were validated with 16S sequencing and IHC. Direct modulation of intratumoral bacteria by local injection of antibiotics into MC38 tumors reduced mast cell levels, while conversely, local antibody blockade of mast cells led to a dramatic increase in microbes, demonstrating an interplay between the host and invading microbes. Asking whether these changes affect ICB therapy, direct intratumoral injection (mostly at the tumor border) of antibiotics in two immunotherapy-responsive tumors (MC-38 and E0771) reduced ICB responsiveness, suggesting that intratumoral microbes were important for ICB responses. Further, in a pancreatic tumor (KPC) that is inherently not responsive to immunotherapy, increasing the microbial burden by depleting mast cells led to immunotherapy responsiveness, which was abrogated by antibiotic treatment, indicating that the intratumoral microbes increased by mast cell depletion were driving the response. Overall, this work highlights the direct relevance of tumor-resident microbes to immunotherapy outcomes.
Microenvironmental metabolites shaping T cell fate and function in cancer - Greg M. Delgoffe - University of Pittsburgh, Pittsburgh, PA
Diverging from his planned talk, Greg Delgoffe presented some new and exciting work on the metabolic features of the tumor microenvironment (TME) and their relationship to immune suppression. Tumor cells alter their metabolism, which in turn affects the metabolic composition of their immediate environment, the interstitial fluid of the TME. The long-known examples include higher oxygen consumption, leading to hypoxia and buildup of reactive oxygen species, and the higher glycolytic rate, leading to hypoglycemia and lactate buildup. These changes have been shown by multiple investigators to affect local T cell biology – CD8+ T cells become exhausted due to hypoxia, and CD4+ T cells differentiate towards regulatory cells as a result of reduced glucose. Lacking though, have been studies that directly link specific metabolite changes to T cell changes in the TME. Delgoffe and his collaborators approached this need by quantitatively characterizing the metabolomic composition of the tumor interstitial fluid (TIF) from murine KPC (KrasG12Dp53f/fPdx1cre) tumors, generating a chemically defined media based on TIF (which could be very specifically modified), and directly analyzing T cell phenotypes in response to such changes. Initial experiments showed that T cells activated in normal media (reflecting the normal periphery), but transferred to TIF media (reflecting the tumor) showed minor inhibition if restimulated soon after transfer. However, prolonged exposure to TIF media before restimulation gradually upregulated PD-1, reduced multi-cytokine production, and dramatically slowed cell growth, even when the T cells were restimulated in normal media, indicating a stable change. To identify what might be missing in TIF media, quantitative comparison of TIF media to plasma identified multiple depleted metabolites. Add-back experiments identified arginine as a key metabolite able to rescue the T cell PD-1 and proliferation phenotypes, but not multi-cytokine production. Turning then to what was built up in TIF media relative to plasma, the researchers identified multiple interesting candidates, but focused the investigation on phosphoethanolamine (pEtn), a metabolite on the path from ethanolamine to phosphatidylethanolamine (PE), an abundant lipid component of cell membranes. Tumor cells frequently downregulate the enzyme ECT/Pcyt2, which is needed for conversion of pEtn ultimately into PE. Remarkably, adding pEtn to T cells in normal media recapitulated the T cell dysfunctional state. Detailed analysis traced the metabolic impact on T cells to a rewiring of T cell metabolism to become addicted to pEtn. Adding other metabolites of the pathway from ethanolamine to PE into normal media also recapitulated T cell dysfunction, pointing to the excess PE produced in these cells as the root cause of the dysfunction. Ultimately, the metabolically rewired T cells used up diacylglycerol (DAG) to metabolize pEtn to PE, and therefore depleted this critical second messenger required for the T cell receptor signaling pathway, resulting in T cell dysfunction. Restoring expression of pyct2 in tumor cells (to eliminate the buildup of pEtn in the media) restored full T cell function and caused reduced tumor size. As a more tractable therapeutic approach, converting the excess PE in T cells to phosphatidylcholine using the liver enzyme PEMT similarly rescued T cell function. As an aside and a general cautionary note, PMA, frequently used to stimulate T cells, contains a DAG analog, and so stimulation with PMA may mask metabolically induced T cell dysfunction.
T cells - Biology and Therapy
Agonistic anti-CD40 converts regulatory T cells in to Type 1 effector cells within the tumor microenvironment - Katelyn T. Byrne - Brenden-Colson Center for Pancreatic Care, Oregon Health and Science University, Portland, OR.
In order to better understand why some patients respond to immunotherapies, while others do not, Katelyn Byrne and colleagues investigated the role of barriers to immunotherapy in mouse models of pancreatic cancer. KP pancreatic tumors models showed high Treg infiltration, even at the early stages of development, and Treg infiltration correlated with total Tregs in PDAC tumors. Using an immunotherapy-responsive model, Byrne evaluated agonist anti-CD40, which can overcome the suppressive TME when used in combination with chemotherapies and/or immune checkpoint blockades (anti-PD-1 plus anti-CTLA-4). In mice with established tumors treated with anti-PD-1 plus anti-CTLA-4, followed by agonist anti-CD40, the researchers noted a rapid reduction of Tregs, and redistribution to the tumor border, while conventional CD4+ T cells and CD8+ T cells remained dispersed throughout the tumor. After confirming that this effect was indeed due to agonist anti-CD40 and not due to immune checkpoint blockade, the researchers began investigating the mechanism underlying this phenomenon. As Tregs do not express CD40, the team hypothesized that antigen-presenting cells likely played a role, and found that DCs and MHC-II were required for anti-CD40-mediated Treg loss. Further, Treg loss required IL-12p40, IFNγ, and host expression of the IFNγ receptor. Next, the researchers questioned whether Tregs were dying or transforming in the TME. To investigate Treg transformation, the researchers traced Foxp3 and Tbet in tumor-bearing mice treated with dual checkpoint blockade and agonist anti-CD40 and observed an increased population of ExTregs, which lost expression of Foxp3 following treatment. Tracking both Foxp3 and Tbet expression, the researchers further found that anti-CD40-induced ExTregs acquired IFNγ effector function and showed evidence of increased NFAT signaling after therapy, indicative of increased TCR ligation. In samples from patients treated with agonist CD40 taken 12 days after treatment, Byrne and colleagues observed patterns similar to those observed in mice, with reduced Tregs and increased Tbet+ Tregs within tumors, and increased Tregs and Tbet+ Tregs in the border regions. Together, these results suggest that agonist anti-CD40 overcomes Treg-mediated immune resistance by inducing changes in APCs that drive Treg reprogramming into Th1-like ExTregs in pancreatic tumors in both patients and mice. With these results, Byrne noted the importance of reconsidering the pool of tumor-reactive T cells present in TMEs after agonist anti-CD40, and the potential for leveraging Treg plasticity to improve patient outcomes.
TCR-gene therapy targeting driver mutations - Eric Tran - Providence Cancer Institute, Portland, OR
TCRs targeting driver mutations may be particularly advantageous in TCR-engineered T cell therapies, given the clonality of driver mutations in tumors and the ongoing requirement for many drivers to maintain tumor viability and/or growth advantage. Eric Tran presented updated data on a patient with pancreatic ductal adenocarcinoma with metastasis to the lung treated with TCR-engineered T cells specific for the G12D variant of KRAS. The patient’s T cells were engineered with a combination of two TCRs specific for Kras-mutant 9mer and 10mer peptides bound to HLA-C-0802 and expanded in the presence of IL-2, IL-7, and IL-21, as well as TGFβ, to attempt to induce a tissue-resident phenotype. The T cell product was a 1:2 mixture of T cells carrying TCRs specific for the 9mer and 10mer peptides. Prior to infusion, the patient received an anti-IL-6 antibody to reduce the severity of cytokine release syndrome (CRS), standard lymphodepletion, and 5 of 6 planned doses of IL-2 support. At 6 months post T cell infusion, the patient showed a 72% RECIST reduction in lung lesions, and the engineered T cells comprised about 2.5% of all circulating T cells in the patient. The response was durable out to a year, but the patient then slowly progressed, and lung lesions required further treatment. For the second cycle of engineered T cell therapy, the patient was treated biweekly with anti-PD-1 starting one day prior to T cell infusion, and only T cells carrying the 9mer-specific TCR were utilized, based on additional in vitro data indicating the higher avidity of this TCR. Interestingly, the patient experienced grade 2 Immune-effector Cell Associated Neurotoxicities (ICANs) beginning on day 4 after infusion, which was not observed for the initial infusion. The ICANs resolved by day 9. To understand the cause of the ICANs, cytokine analysis of serum samples collected daily after the first and second infusion showed very similar kinetics for some cytokines, and small differences for others, but no clear indication of a relationship to the ICANs. IL-1β, which has been associated with neurotoxicity in some models, only showed a difference in level after the ICANs resolved. IL-8 levels were increased during the first 5 days after the second infusion compared to the first. IL-8, also known as CXCL8, can promote chemotaxis and degranulation of neutrophils. CD30, an immunostimulatory receptor, was also elevated specifically during the time of ICANs, which may be of significance. Clinically, the patient’s tumors were stabilized (some clearly shrank), which has been maintained at 6 months by RECIST, with some suggestion of progression. The engineered T cells after the second infusion showed lower persistence (about 0.6% at 6 months) than after the first. To address why the patient regressed after the first infusion, flow cytometry analysis of a tumor resected at progression showed that the engineered T cells were still present in the tumor, with expression of markers such as TIGIT, TIM3, IL-7Rα, and CD25, indicative of antigen experience. Immunohistochemistry of an excised, PET-avid lesion showed a close relationship between CD3+ T cells and CD163+ macrophages. A separate, non-PET-avid lesion identified the presence of neutrophils near large tumor cells, and the tumor cells appeared to be less viable. Further analysis will be needed to determine the role, if any, of these neutrophils. In sum, understanding the causes for recurrence, toxicity, and further response in this patient may provide important insights for future TCR-engineered T cell therapy.
Dissecting mechanisms of escape from T cell attack in cancer stem cells - Judith Agudo - Dana-Farber Cancer Institute, Boston, MA
To tackle the question of whether tissue stem cells are really immune privileged, Judith Agudo developed JEDI (just EGFP death-inducing) T cells that target GFP-expressing cells. By treating Lgr5-GFP mice with JEDI T cells, Agudo showed that Lgr5+GFP+ stem cells in the skin are indeed immune privileged and escape T cell-mediated killing via downregulation of MHC class I. In the intestine, however, Lgr5+GFP+ stem cells were highly immunogenic and were easily eradicated by JEDI T cells. In contrast, DNA repair-proficient colorectal cancer is highly resistant to immunotherapy, leading Agudo to question how cold colorectal cancers can form from these highly immunogenic intestinal cancer stem cells and evade immune surveillance. Colonoscopy-guided implantation of colon organoids harboring mutant KrasG12D with loss of p53 and APC (AKP) into the colon of recipient mice, and several passages, created tumor-derived AKP organoids, in which Sox17 was found to be upregulated by RNAseq and ATACseq. Sox17 is a transcriptional factor involved in the ventral foregut formation during fetal development, and while it is silenced in healthy colon tissue, it is essential for colon cancer development. Sox17 knockout showed drastic reduction in tumor growth in immunocompetent mice, but not in immunodeficient mice, suggesting a potential role of Sox17 in immune evasion. Single cell RNAseq, immunohistochemistry, and FACS analyses showed an increased infiltration of effector-like IFNγ+ CD8+ T cells and almost no exhausted CD8+ T cells in Sox17 KO compared to wild-type tumors. CD8+ T cell depletion rescued growth of Sox17 KO tumors. Furthermore, Sox17 loss induced enrichment of interferon gene signatures, increased IFNγ signaling via upregulation of IFNAR1, and MHC I in colon organoid cultures. More experiments showed that Sox17 directly repressed the IFN receptor expression in these organoids, and IFNGR1 deletion rescued the tumor growth in Sox17 KO tumors. Since colon cancers originate from adenomas, the researchers crossed Lgr5-CreERT-GFP mice with APCf/f mice to investigate the role of Sox17 in adenoma formation and showed that Sox17 is highly expressed in adenomas, but is mutually exclusive with Lgr5. Loss of Sox17 prevented the formation of adenomas, in a CD8+ T cell-dependent manner. In adenomas, Sox17 repressed Lgr5 expression and Sox17 deletion expanded Lgr5+ MHC class I-expressing cells. In summary, induction of a fetal program in intestinal stem cells upregulates Sox17, which in turn downregulates IFNAR1, IFNγ signaling, and MHC class I to evade immune surveillance. Once the tumor mass and an immunosuppressive tumor microenvironment has formed, Lgr5 is turned back on in some cells, allowing them to form metastases and contribute to tumor recurrence.
Tumor-associated lymphatic vessels and T cell transit through the tumor microenvironment - Amanda W. Lund - NYU Langone Health Perlmutter Cancer Ctr., New York, NY
Covering an often overlooked feature of the tumor microenvironment, Amanda Lund discussed the relevance of lymphatic vessels in tumors, which flow unidirectionally towards lymph nodes and can respond dynamically to signals in their environments (e.g., VEGFA, IFNγ), allowing them play diverse roles in tumors, from tumor metastasis to immune mediation and control of stem cell niches. In recent work, Lund and colleagues investigated how lymphatic vessels regulate on/off signals that drive immune responses to cancer. Previous work showed that in mice bearing melanoma, the absence of dermal lymphatic vessels improved immune responses to adoptive T cell transfer. When T cells were tracked in repeat experiments, it became evident that while T cell activation and initial infiltration were similar regardless of lymphatic vasculature, the accumulation of tumor-specific CD8+ T cells was increased at tumor sites at later time points. Interestingly this was not due to increased proliferation, as T cells at these tumor sites were actually less proliferative. Lund hypothesized that the lack of lymphatic vasculature was limiting T cell egress, keeping both tumor-specific and bystander T cells at the tumor site for longer periods of time. Investigating T cell egress in mice with functional lymphatic vasculature, Lund and colleagues used a photoconvertible mouse model. Photoconverting tumors and endogenous cells in the tumor from green to red allowed the researchers to track any newly infiltrating or egressing cells. These studies revealed that 24 hours after photoconversion, high numbers of T cells, including a high portion of CD8+ T cells, had egressed from tumors and ended up in draining lymph nodes. Tracking tumor specificity, the researchers found that egressing T cells included many functional tumor-specific T cells with the capacity for further activation, suggesting that lymphatic vasculature mediates the early exit of desirable antitumor effectors. Exploring what factors define which T cells stay or leave, Lund and team compared retained and egressing cells and found that the chemokine receptor repertoire is tuned by intratumoral T cell states, with non-activated T cells enriched in CXCR4 and more likely to leave, and activated/antigen-experienced T cells enriched in CXCR6 and more likely to stay. In line with this, adoptive transfer of CXCR4-deficient CD8+ T cells impaired T cell exit and improved tumor control. These results support the notion of a “tuning” of T cell migratory potential depending on whether a T cell has encountered its antigen. Looking again at the role of the lymphatic vessels, Lund showed that inflamed lymphatic vessels, like those in tumors, express CXCL12 (a ligand for CXCR4), luring non-activated CD8+ T cells from tumors. This was confirmed with knockout of CXCL12, which induced dramatic redistribution of CD8+ T cells from the periphery of tumors into the centers of tumor, enhancing CD8+ T cell-mediated tumor control. Lund hopes to further investigate these mechanisms and potentially exploit them for immunotherapies that could enhance the retention of tumor-specific effector T cells within tumors.
Safety and efficacy from the phase 1/2 first-in-human study of REGN5459, a BCMAxCD3 bispecific antibody with low CD3 affinity, in patients with relapsed/refractory multiple myeloma - Attaya Suvannasankha - Indiana University Simon Cancer Center and Roudebush VAMC, Indianapolis, IN
To provide a treatment option for patients with multiple myeloma, who inevitably relapse after multiple prior therapies, Attaya Suvannasankhha presented the results of a phase I, dose-escalating trial of a novel bispecific T cell-engaging antibody REGN5459, binding to BCMA and CD3. This fully human antibody (derived from Veloci-Bi™ mice) was engineered to have a lower affinity to CD3 than other typical T cell engagers. In preclinical models, REGN5459 activated T cells and depleted plasma cells with low cytokine release. All 43 patients on the trial were heavily pretreated and had no other available options. Initial step-up dosing over the first two or three weeks was investigated to mitigate cytokine release syndrome (CRS), often seen with other bispecifics, followed by weekly dosing until week 16, and then biweekly dosing. Patients were pre-medicated with dexamethasone and optional acetaminophen and/or antihistamine. When the full dose was well tolerated, pre-medication was tapered and then stopped. The dose escalation phase identified a recommended phase 2 dose (RP2D) of 480 mg for an expansion cohort to assess overall response. A maximum tolerated dose (MTD) was not reached. Infections occurred in about 63% of patients, including high-grade infections in about 30% of patients. CRS was the most common treatment-related adverse event, which increased in frequency, but not severity, with dose. 16% of patients discontinued treatment due to adverse events. Response rates increased with dose, and at the highest doses (the RP2D and one patient at 900 mg), 91% of patients responded, with 62% complete responses. Responses occurred early, with a median time to response of 0.8 months. A median duration of response was not reached at a median follow-up of 9 months, with the longest responses being 26+ months as of data cut-off.
Checkpoint blockade and combination therapies
Genomic landscape of response and resistance to immune checkpoint blockade - Valsamo K. Anagnostou - Johns Hopkins University, Baltimore, MD
Focusing on tumor-intrinsic features that impact responses to immunotherapy, Valsamo Anagnostou began by pointing out that multiple studies have shown that high tumor mutation burden is associated with response to immune checkpoint blockade, which is exemplified in the context of microsatellite instability. However, several technical and biological factors pose challenges (tumor sample purity, assay harmonization, other contributing features) in using mutational burden as a biomarker for response to ICB. Integrative exome and transcriptome analysis, however, can provide better outcomes in predicting response to ICB. For example, multi-omics meta-analysis showed that specific genomic features are associated with response in a pan-cancer manner, while some are cancer type- and lineage-specific. Tumors that regressed with chemoimmunotherapy harbored a higher degree of chromosomal imbalance, suggesting that chromosomal deletion results in an ability to eliminate mutations in haploid regions, and as such, these mutations persist and are associated with response to ICB. Valsamo and colleagues thus hypothesized that there are subsets of mutations (“persistent mutations”) that are biologically distinct and are unlikely to be deleted via chromosomal deletion (which would be a lethal event for the cell), and thus result in sustained immunological tumor control. These persistent mutations can be single-copy mutations in haploid regions or multi-copy mutations, and differ from single-copy mutations in diploid regions, which are “loss-prone”. In the case of lung cancer, a subset of candidate mutation-associated neoantigens present in the baseline tumors were eliminated in ICB-resistant tumors, and potentially drove the emergence of resistance. The neoantigen loss occurred either via immune elimination of neoantigen-containing cells or via genetic events leading to neoantigen loss and expansion of the resistant clone. Indeed, in lung cancer examples, several neoantigen mutations were eliminated by genetic events, and these were confirmed to be immunogenic, consistent with these genetic events leading to tumor resistance. Quantifying persistent tumor mutation burden (pTMB; one-copy or multi-copy persistent mutations) revealed that the fraction of persistent mutations and the relative contribution of multi-copy versus one-copy mutations depended on cancer lineage, and tracked distinctly from the overall tumor mutation burden. In fact, many tumor types changed classification (low vs high mutation burden) depending on whether one calculated overall TMB or pTMB. Tumor aneuploidy and persistent mutations showed a range of relationships across tumor types, and there were very few persistent mutations in driver genes, except for tp53. Persistent mutation burden better distinguished responding patients compared to overall mutational burden, loss-prone mutation burden, or tumor aneuploidy. Persistent mutations persist through tumor evolution, and tumors with higher pTMB are more inflamed and associated with cytolytic activity and immunotherapy response. In summary, Anagnostou noted that understanding the genomic wiring of immune oncology responses is key, and can inform patient selection strategies. TMB does not fully capture tumor foreignness, and persistent mutations represent an un-editable set within the overall TMB, which are not eliminated by chromosomal loss or therapeutic selective pressure. Persistent mutations may thus mediate more sustained neoantigen-driven immune responses, resulting in long-term clinical benefit with immunotherapy.
Intratumoral (IT) MEDI1191 + durvalumab (D): Update on the first-in-human study in advanced solid tumors - Eduardo Castañón - Clínica Universidad de Navarra, Pamplona, Spain
A lipoparticle-formulated mRNA encoding IL-12, termed MEDI1191, can promote local IL-12 production and enhance innate and adaptive CD8+ T cell-dependent antitumor immunity upon intratumoral (i.t.) injection in murine models. Eduardo Castañón presented results from a first-in-human phase 1 clinical trial investigating the combination of i.t. MEDI1191 and intravenous (i.v.) anti-PD-1 (durvalumab) in heavily pre-treated patients with advanced solid tumors. A total of 61 patients were enrolled on the trial. In the dose-escalation part 1A, patients with cutaneous or subcutaneous lesions received MEDI1191 on day 1 and 22, and thereafter, durvalumab every 4 weeks. In parts 1B (patients with cutaneous or subcutaneous lesions) and 1D (patients with deep-seated lesions and, in all cases, liver metastases), patients received concurrent MEDI1191 and durvalumab on days 1, 29 and 57, and thereafter, durvalumab every 4 weeks and MEDI1191 every 8 weeks. Treatment was continued for up to two years or until disease progression or intolerable toxicity. No dose-limiting toxicity or maximum-tolerated dose was observed. The most common adverse events were fatigue and pyrexia, and the most common grade 3 adverse event was anemia. Antitumor activity was observed in injected and uninjected local or distant lesions, confirming an abscopal effect. Responses were evaluated at an uninjected target lesion. The overall response rate was 8%, with 5 confirmed partial responses (PR), including in 3 patients previously treated with anti-PD-(L)1 and/or anti-CTLA-4 therapy. 25% of patients had stable disease (SD), including 2 patients with an unconfirmed PR. Responses lasted for up to 22 months, and the median duration of response has not been reached. Across dose cohorts, increases in serum IL-12 peaked at 24 hours after injection, and increases in IFNγ, CXCL9, CXCL10, and CXCL11 levels were also observed 24 hrs after the first and second dose of MEDI1191 or MEDI1191 plus durvalumab. However, none of the increases in cytokines or chemokines were dose-related to MEDI1191. Paired biopsies revealed enhanced tumoral PD-L1 expression and increased tumor infiltration of CD8+ and CD3+Ki67+ cells in about 40-50% of the patients. Patients with PR/SD showed a trend towards a greater increase in the expression of key antitumoral gene signatures compared to patients with progressive disease.
The contribution of agonists to combination immunotherapy for PD-1 resistant cancers - Elizabeth M. Jaffee - John Hopkins University, Baltimore, MD
While immunotherapy has made great strides against certain cancers, most are still resistant, even when infiltrated with T cells. Investigating the quality of T cell responses in such tumors, Elizabeth Jaffee and colleagues found that these T cells are not great effectors, and hypothesized that inducing higher quality effector T cell responses could enhance antitumor immunity. Investigating TCR avidity as a potential feature of T cell quality, Jaffee found that in a non-tolerized mouse model, responses from T cells with low avidity TCRs could be enhanced with vaccination, while responses from T cells with high avidity T cells could sufficiently clear tumors on their own. In tolerized mice, low avidity TCRs were insufficient, even with the vaccine, and only high avidity TCRs with vaccination induced T cell trafficking and tumor clearance. However, IFNγ production and tumor clearance could be induced with the addition of an OX40 or 4-1BB agonist to T cells with low avidity TCRs plus vaccination. More recently, Jaffee and others evaluated patient samples from neoadjuvant/adjuvant clinical trials. In the first arm, treatment consisted of vaccine alone, which induced infiltration of lymphoid aggregates showing signs of T cell activation and monocyte PD-L1 upregulation. Patients saw significant improvements in disease-free and overall survival, which could be associated with expansion of mesothelin-specific CD8+ T cells and increased T cell avidity. Upon deeper investigation, the researchers found that both CD4+ and CD8+ T cells were increased in tumors, but were not activated. Based on these results, the researchers added anti-PD-1 in the second arm. In this setting, the lymphoid aggregates, which were likely tertiary lymphoid aggregates, were actively forming and educating both T and B cells. Th17 T cells (which may arise from reprogramming of Tregs) were also found leaving these aggregates and colocalizing with tumor cells. In patients with long-term survival, there was an increase in the Th1:Th2 ratio, and a reduction in exhausted CD4+ and CD8+ T cells in tumors. Adding anti-PD-1 increased antigen-presenting monocytes and reduced inhibitory monocytes, suggestive of macrophage reprogramming. It was also noted that anti-PD-1 increased Tregs, which were associated with worse prognosis when colocalized with tumor cells. In both arms, pre-treatment neutrophils (which express IL-8R) were associated with worse prognosis. In post-treatment samples, higher levels of neutrophils were associated with CD4+ and CD8+ T cell exhaustion and shorter patient survival. Also in both groups, an increased CD8+ T cell population expressing CD137 was associated with granzyme B expression and longer survival. To enhance this population, the researchers tested the addition of agonist anti-CD137 to vaccine plus anti-PD-1. In a mouse model of pancreatic cancer, this triple combination was the only treatment to reduce the disease burden. In patients, triple combination proved to be the first treatment in this study to induce pathologic responses, with 3 of 10 patients achieving near complete responses. Median disease-free survival has not yet been met, with many of the patients two years out. Looking at immune response in patients from each arm, total CD8+ T cells went up progressively with each arm. Tregs, on the other hand, had increased in the second arm, but decreased in the third arm with the addition of agonist anti-CD137. Clonal expansion also increased with each arm, with a high portion of hyperexpanded clones observed in the third arm. Markers of trafficking, exhaustion, and cytoskeletal remodeling increased in the second arm, and cytoskeletal remodeling and chemokines/trafficking were further enriched in the third arm, suggestive of increased motility of infiltrating T cells.
Myeloid cells and DC therapy
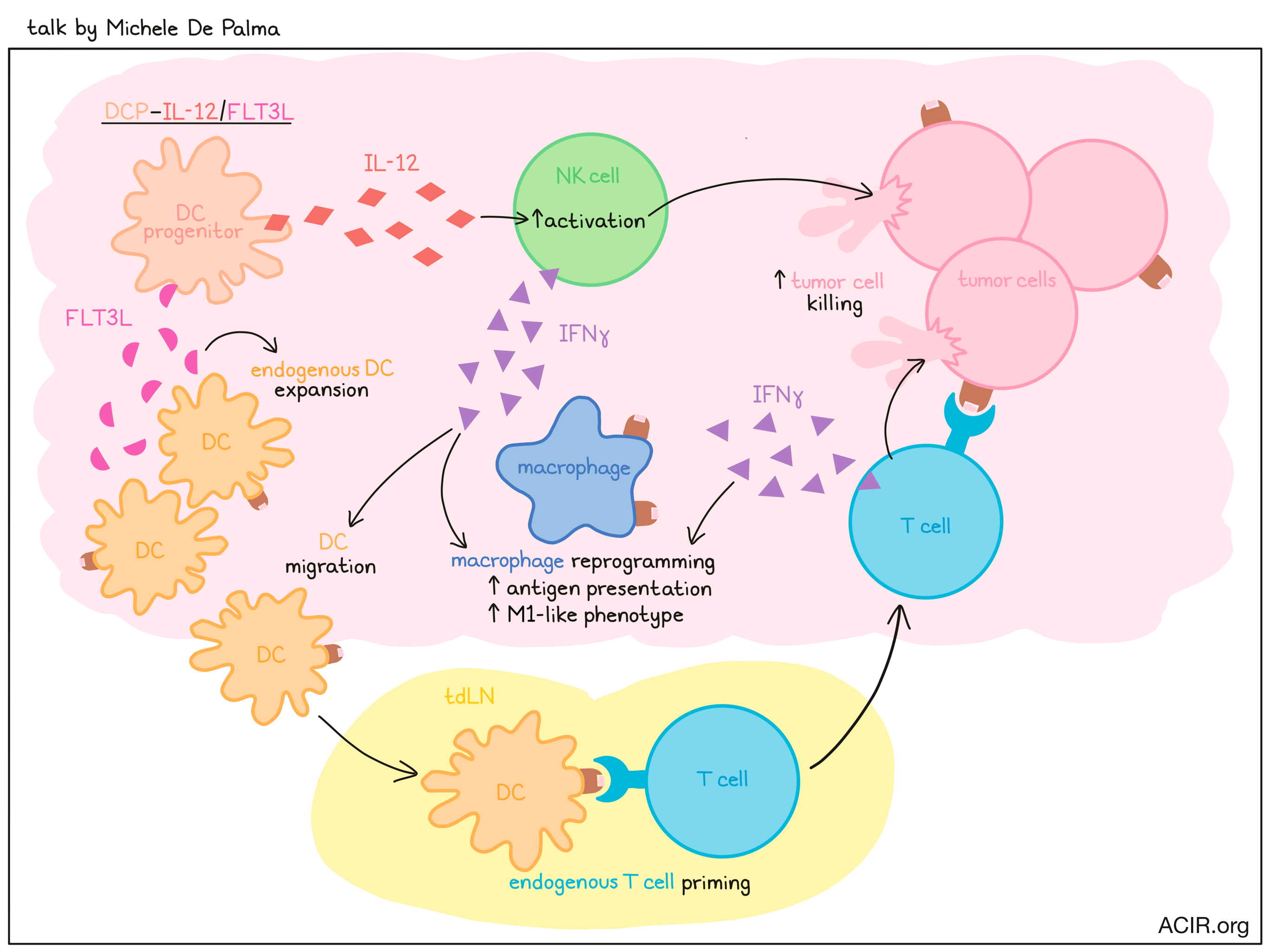
Antigen-agnostic dendritic cell therapy - Michele De Palma - The Swiss Federal Institute of Technology Lausanne (EPFL), Lausanne, Switzerland
Dendritic cell vaccines are usually made with monocyte-derived dendritic cells pulsed with tumor lysate or tumor-associated antigens, but the clinical efficacy of these types of vaccines has often fallen short of expectations. Michele De Palma hypothesized that this could be related to incomplete identification of antigens and/or to the use of monocyte-derived DCs, which are easy to generate from monocytic progenitor cells, but lack the cross-presentation capacities of professional antigen-presenting cells like cDC1s, which are derived from a common DC progenitor (DCP). Hypothesizing that DCPs might serve as a better source for therapeutic DCs, De Palma developed a method for generating DCPs from hematopoietic stem and progenitor cells (HSPCs) using a cocktail of cytokines. Following adoptive transfer into mice, DCPs matured exclusively into immature DCs, cDC1s, and cDC2s, and engrafted more efficiently into spleens than monocyte-derived DCs. Looking at total cDC1 and cDC2 populations in these mice, including endogenous cells, the researchers found that up to 50% of cDC1 were derived from graft, suggesting that DCPs could support DC reconstitution, even without conditioning. Exploring strategies to further enhance DC functionality in vivo, De Palma and colleagues engineered DCPs to produce FLT3L, which supports DC development, and/or IL-12, which helps to induce T cell immunity. Upon adoptive transfer, increased levels of circulating FLT3L and IL-12 could be detected for a brief window of time before DC differentiation or death. In mice transplanted with B16F10 melanoma cells, DCPs expressing FLT3L alone induced no tumor control, DCs expressing IL-12 induced some tumor control, and the combination induced potent antitumor control that prevented the development of viable tumors. In line with this, IL-12 expression marginally increased CD4+ and CD8+ T cell infiltration into tumors, whereas the combination with FLT3L expression showed clear synergy in regards to increased T cell infiltration and IFNγ production. Further, scRNAseq revealed conserved type I and II IFN responses in both melanoma and myeloid cells, suggesting that DCP-IL-12/FLT3L broadly activated IFNγ signaling in the tumor microenvironment. Further investigation into the mechanism behind this antigen-agnostic antitumor effect revealed that DCP-IL-12/FLT3L induced expansion of endogenous DCs through FLT3L and activation of NK cells through IL-12. The activated NK cells produce IFNγ, which supports DC migration to the tumor-draining lymph node, where DCs further expand and prime endogenous T cells. Primed and activated T cells then travel to the tumor, where they induce antitumor immunity and contribute additional IFNγ, which supports a shift in macrophages towards an antigen-presenting M1-like phenotype. Monocyte-derived DCs engineered to express FLT3L and IL-12 were less effective. Finally, De Palma described an experiment in which DCP-IL-12/FLT3L were administered in combination with GD2-CAR T cells into mice bearing GD2-expressing gliomas. While both the DCP-IL-12/FLT3L cells and GD2-CAR T cells were ineffective as monotherapies, their combination induced robust antitumor responses, with many mice experiencing complete regressions and survival with no detectable glioma, even with imaging at a cellular level. With these impressive in vivo results, De Palma is hopeful that the success of this strategy can be translated to the clinic.
Basic and clinical impacts of targeting myeloid cells in tumors - Judith A. Varner - UCSD Moores Cancer Center, La Jolla, CA
Macrophages are associated with tumor progression and resistance to therapy, and can comprise up to 30-60% of cells in a tumor. Judith Varner suggested two options for targeting TAMs: inhibit the recruitment of bone marrow-derived macrophages to the tumor, or repolarize macrophages towards the pro-inflammatory M1 phenotype. Varner and her team identified PI3Kγ for its predominant expression in myeloid cells and its role in myeloid cell recruitment and polarization during inflammation. Inhibition of PI3Kγ promotes polarization of macrophages towards an M1 phenotype, with an increase in inflammation- and antigen presentation-related genes and a decrease in the expression of genes encoding immunosuppressant factors. Mechanistically, PI3Kγ inhibition changed chromatin accessibility and altered transcription in macrophages. Therapeutic inhibition of PI3Kγ reduced tumor growth and metastasis in a number of tumor models, but the effect varied based on the tumor microenvironment. Inflamed tumors showed a better response, with an upregulation of gene signatures related to T cell activation, innate immunity, and antigen presentation, and enhanced MHC class II expression on tumor-associated macrophages. Single-cell analysis of T cell non-inflamed Lewis lung carcinoma, focusing on myeloid cells, confirmed higher MHC class I and II molecule expression in PI3Kγ KO compared to wild-type mice. In an HPV+ head and neck tumor model, PI3Kγ inhibition synergized with anti-PD-1 therapy, increasing long-term survival. Furthermore, the researchers observed a synergistic increase in intratumoral CD8+ T cells, granzyme B, IFNγ, and IL-12 expression, and a decrease in CD4+ T cells. Single-cell analysis revealed a shift in the myeloid cell population towards a pro-inflammatory phenotype, with an increase in IFNγ responses and MHC-related genes in PI3Kγ KO compared to wild-type tumors. PI3Kγ inhibition increased the number of activated effector and effector memory CD8+ T cells, and activated NK cells in the tumor, promoted T cell recruitment, relieved exhaustion, and promoted TCR diversity. The antitumor effect of PI3Kγ inhibition was CD8+ T cell-, IL-12-, and IFNγ-dependent. In combination with checkpoint inhibitors, PI3Kγ inhibition promoted long-term persistent immunological memory, with increases in effector memory T cells in the spleens of treated animals. In a clinical trial, IPI-549 (eganelisib), a first-in-class oral PI3Kγ-selective inhibitor, in combination with nivolumab as a second-line therapy, demonstrated a 42% reduction in the risk of death at 2 years in patients with advanced platinum-refractory urothelial cancer. The responding patients showed an enrichment in gene sets associated with antigen presentation, IFNγ signaling, and TCR signaling in peripheral blood, consistent with the findings in tumor models. Varner and colleagues also identified the Treg biomarker integrin beta 8 (ITGB8) as a resistance factor. ITGB8 was highly expressed in CD4+ T cells in patients treated with the combination therapy, and inhibition of ITGB8 and PI3Kγ demonstrated synergistic antitumor efficacy in animal models.
Back to Top
By Ute Burkhardt, Ed Fritsch, Lauren Hitchings, and Shishir Pant



