Previous research has shown that the immune system contributes to chemotherapy response; therefore, immunosuppressive mechanisms can be hypothesized to limit the cytotoxic effect. In a study recently published in Nature Cell Biology, Salvagno et al. demonstrated that targeting immunosuppressive macrophages and neutrophils enhances the efficacy of platinum-based chemotherapy in a mouse breast cancer model.
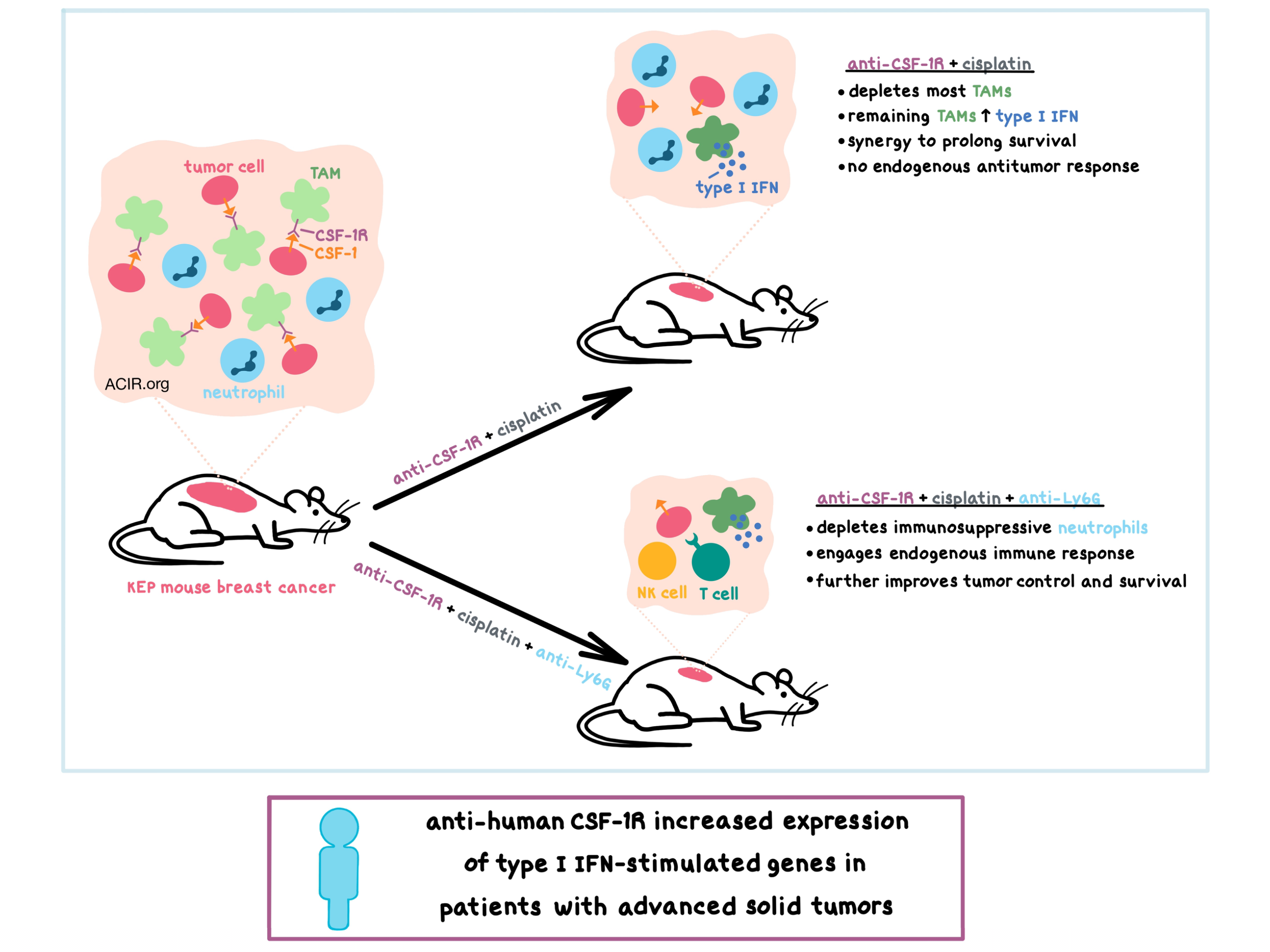
To find ways to enhance the efficacy of chemotherapy, researchers utilized the poorly immunogenic K14cre;Cdh1F/F;Trp53F/F (KEP) transgenic mouse model of spontaneous mammary tumorigenesis, which is similar to human invasive lobular carcinoma. They found that both cancer and host cells in KEP tumors express high levels of colony-stimulating factor-1 (CSF-1), which leads to high infiltration by macrophages. The CSF-1 receptor (CSF-1R) was found to be expressed on tumor-associated macrophages (TAMs) at high levels and on infiltrating monocytes and neutrophils at lower levels. Treating tumor-bearing KEP mice with anti-CSF-1R significantly reduced the TAM population, however, it did not affect tumor growth or metastasis formation. Anti-CSF-1R synergized in combination with cisplatin or oxaliplatin (platinum-based, DNA cross-linking chemotherapeutic agents) to improve tumor control and prolong survival compared to chemotherapy with a control antibody. Interestingly, such synergy did not occur between anti-CSF-1R and docetaxel (an antimitotic chemotherapeutic agent).
While anti-CSF-1R depleted the majority of TAMs, which were CD11bhi CD206lo with some expression of MHC-II, the CD11b+F4/80+ cells that remained in the tumor appeared to have an altered profile – they had increased expression of MHC-II, PD-L1, monocyte marker Ly6C, and costimulatory molecules CD80 and CD86, and decreased expression of chemokine receptors CCR2 and CX3CR1. The results of further experiments suggested that this cell population may have arisen from newly recruited circulating monocytes, which were increased in the tumors after anti-CSF-1R treatment.
Digging deeper to understand the mechanism behind the synergistic response of the anti-CSF-1R and cisplatin combination therapy, the researchers utilized RNAseq and found that the CD11b+F4/80+ cells remaining in the tumor after combination treatment were strongly enriched in type I IFN production and signaling genes. This cell population expressed lower levels of CSF-1R, potentially explaining resistance to anti-CSF-1R therapy. Mechanistically, anti-CSF-1R upreguled intracellular pattern recognition receptors that signal to induce type I IFN production, and this increase in intratumoral type I IFN signaling was found to be essential for the antitumor synergy of the anti-CSF-1R and cisplatin combination therapy. Further experiments demonstrated that type I IFNs also directly inhibited KEP cancer cells.
Interestingly, the efficacy of the combination treatment was not dependent on CD8+ T cells or any other components of the adaptive immune system, indicating that the anti-CSF-1R-mediated type I IFN-rich tumor microenvironment was not sufficient to induce an antitumor T cell response. Although the anti-CSF-1R led to enriched type I IFN signaling genes in neutrophils, Salvagno et al. suspected that this cell population was immunosuppressive in the context of the KEP tumors. Adding the neutrophil-depleting anti-Ly6G to anti-CSF-1R and cisplatin further improved tumor control and survival, and this additional benefit was dependent on the innate and adaptive immune response, including NK cells and CD8+ T cells.
Seeking to validate the preclinical results in patients, Salvagno et al. analyzed pre-treatment and on-treatment tumor biopsies from patients with advanced solid tumors who were treated with emactuzumab, a humanized anti-human CSF-1R monoclonal antibody. Concordant with the preclinical data, gene expression profiling of the biopsies indicated that treatment increased the expression of multiple type I IFN-stimulated genes.
Overall, Salvagno et al. demonstrated that in the KEP transgenic mouse model of breast cancer, targeting TAMs with anti-CSF-1R depleted most of the macrophages and altered the profile of the remaining CD11b+F4/80+ cells to induce the production and signaling of type I IFN, which was required for the synergy between anti-CSF-1R and platinum-based chemotherapy and led to improved survival. However, additional depletion of the immunosuppressive intratumoral neutrophils with anti-Ly6G was necessary to engage the endogenous antitumor response in order to further boost tumor control and prolong survival. The ability of anti-human CSF-1R treatment in patients with advanced solid tumors to increase the expression of type I IFN-stimulated genes indicates the potential clinical relevance of this combinatorial approach.
by Anna Scherer



