
Last week, the ACIR team attended the AACR Annual Meeting 2018 in Chicago. This week’s extensive special feature covers select sessions from the conference. We have organized the content by topics below.
Immune Checkpoint Blockade
Neoantigen Targeting
Immune Cell Agonism
Metabolism
Immune Cell Biology
Adoptive Cell Transfer
The AACR Annual Meeting 2018 opened with a plenary session on advancing cancer medicine, from discovery to patient care. Alexander Eggermont presented the first results from the EORTC 1325-MG/KEYNOTE-054 phase III clinical trial, an interim analysis after 351 events that compared pembrolizumab (anti-PD-1) versus placebo in 1019 patients with resected high-risk stage III melanoma with regional positive lymph nodes. At 1 year follow-up, recurrence-free survival was significantly better in the pembrolizumab group (75.4%) than in the placebo group (61.0%), corresponding to a hazard ratio of 0.57; the trend continued to 18 months and beyond. Survival was similar between patients with PD-L1+ and PD-L1- tumors. These results were distinct from the KEYNOTE-001 trial data, which demonstrated increased survival in patients with PD-L1+ tumors. Pembrolizumab-related serious adverse events occurred in 14.7% of patients versus 3.4% in the placebo group, and most events were managed with standard-of-care. However, approximately 20% of patients experienced long-term thyroid-related disorders that were drug-related; most were grade 1-2, although <2% were severe. One patient experienced pembrolizumab-related grade 5 myositis, which contributed to their death. The trial is ongoing, and long-term survival data remains to be determined.
Presenting results of the first interim analysis of Keynote-189, a randomized phase III clinical study (616 patients enrolled) testing the combination of immunotherapeutic pembrolizumab with chemotherapeutic pemetrexed and platinum as a first-line therapy for metastatic non-squamous non-small cell lung cancer (NSCLC), Leena Gandhi revealed that the trial had met its dual primary endpoints of overall and progression-free survival, reducing the risk of death by half. OS or PFS increased with PD-L1 levels but significant benefit was observed even when PD-L1 was less than 1%. Safety was manageable in the combination arm, with a slightly increased rate of renal toxicity. Their results suggest a new standard of care in treatment of metastatic NSCLC.
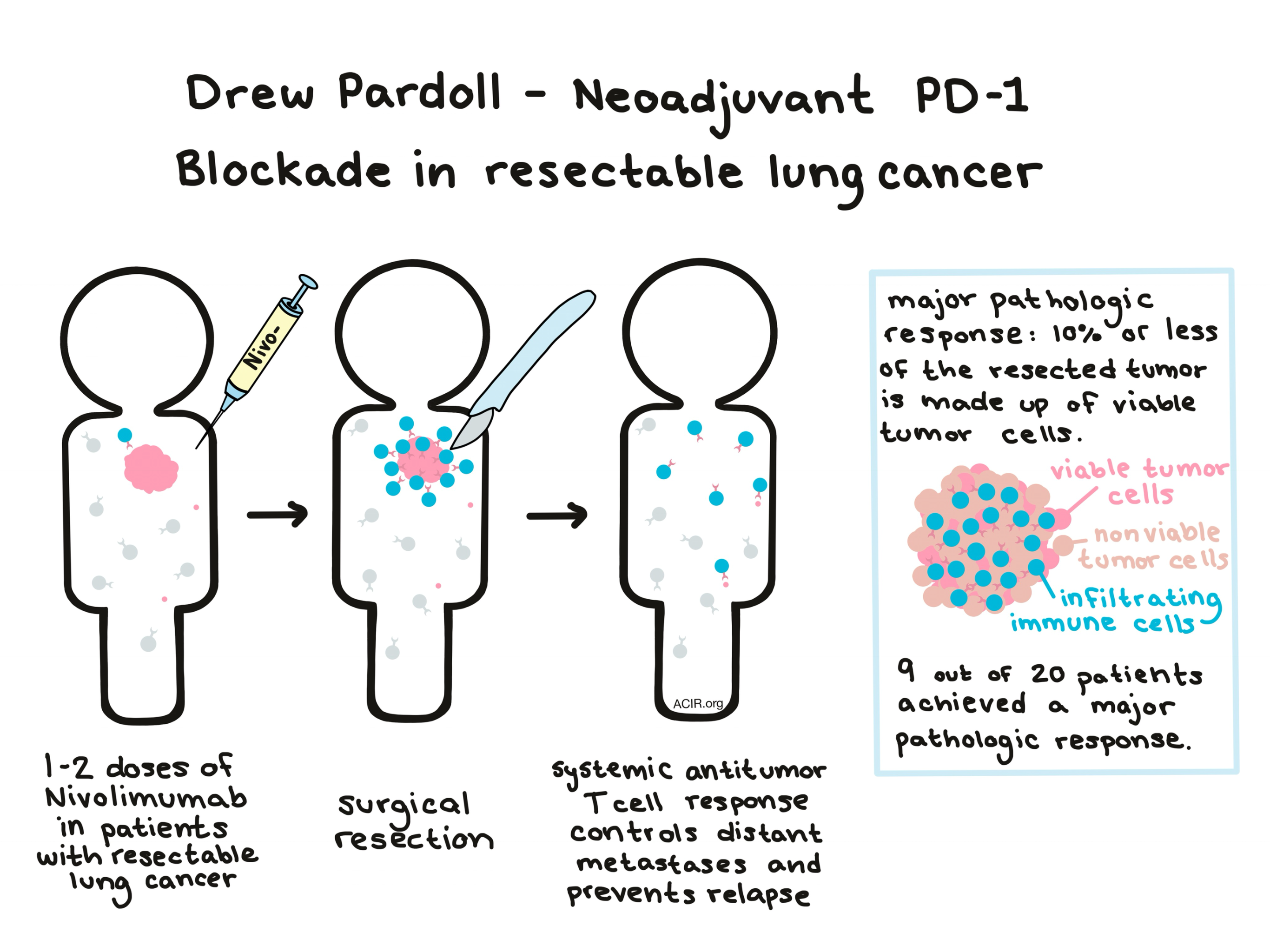
Drew Pardoll presented the results of a phase I clinical trial using Nivolumab prior to surgical resection of stage I-III NSCLC, based on the rationale that neoadjuvant anti-PD-1 treatment would allow the tumor to prime T cells that would circulate systemically and destroy distant micrometastases to prevent relapse after surgery. In addition to showing that nivolumab was safe and did not delay surgery, the trial yielded major pathologic responses (defined as 10% or fewer viable tumor cells in the resected tumor) in nine of twenty patients; three patients had no evidence of viable cells in the resected specimen at all. Pathologic response was associated positively with mutation burden. While it is too early to tell whether these results will ultimately lead to improved progression-free or overall survival, major pathologic responses are promising, and expansion of circulating neoantigen-specific T cells found in the blood suggest systemic antitumor immunity.
Bob Schreiber described an elegant and comprehensive analysis of the transcriptomic and functional marker analysis of tumor-infiltrating myeloid cells following various checkpoint therapies. His model system utilizes the well-studied methylcholanthrene-induced tumor line, T3, which is sensitive to neoantigen vaccination (requiring both CD8 and CD4 epitopes) and checkpoint blockade alone or in combination. Using single cell RNA sequencing and CyTOF analysis of tumors just prior to tumor breakdown following anti-PD-1 and/or anti-CTLA-4 therapy, Schreiber’s team was able to observe dramatic changes in the myeloid compartment as immunosuppressive macrophage-like cells (CXCR3+ and CD206+) disappeared and new populations (Cd1d1+, PD-L1+, iNOS+) emerged. Importantly, these changes depended critically on IFNγ, suggesting a feedback loop with activated T cells. Both checkpoint molecules contributed to the changes, although the relative contributions varied and the two high-dimensional analytical techniques provided both confirmatory and complementary information.
In a session focusing on defining new immunotherapeutic targets through deep molecular characterization, Steven Pai explored how tumor burden affects response to combination anti-PD-1/anti-CTLA-4 treatment. In a TRAMP-C2 mouse model, combination checkpoint therapy (anti-CTLA-4 and anti-PD-1) in the low tumor burden state (immediately after implantation) surprisingly increased the tumor growth significantly. The researchers found that in this setting, combination treatment drives the loss of dominant, activated, antigen-specific T cells by activation induced cell death (AICD) via high IFNγ expression. This is not observed in dual checkpoint treated mice in the high tumor burden state (30 days after implantation), where T cells are more exhausted and produce less IFNγ. Knocking out IFNγ receptor in mice restored response to combination therapy. Overall, these results indicate that combination therapy benefit may vary by disease context.
In the same session, Stephen Mok examined the quality of memory cells in mice treated with either anti-CTLA-4 or anti-PD-1 during initial tumor challenge or during priming with a whole tumor cell vaccine. In mice with melanoma, both therapies elicited initial antitumor response; however, the antitumor response upon rechallenge (>70 days) was robust with anti-CTLA-4 but weak with anti-PD-1. Further analysis of the mechanism showed that anti-PD-1 response was completely dependent on CD8+ T cells, while anti-CTLA-4 response was only partially dependent on CD8+ T cells. There was no difference in the frequency of antigen-specific T cells between the two treatments in the memory phase prior to rechallenge, nor was there a difference in the ratio of central memory/effector T cells at the memory and rechallenge phases of response. However, anti-CTLA-4 treatment resulted in greater expansion of memory T cells during rechallenge and enhanced IFNγ production during the memory phase compared with anti-PD-1 treatment, highlighting mechanistic differences between the two therapies.
In a major symposium focused on personalized neoantigen vaccines, Lelia Delamarre discussed two key challenges in vaccine development: predicting neoantigen presentation and predicting immunogenicity. Addressing presentation, she discussed how her team developed a high-throughput assay to measure the binding affinity of peptides to generate a large, quantitative, predictive dataset of MHC:peptide presentation. She also discussed how they used mass spectrometry to show that interferon gamma treatment upregulated the antigen processing machinery in tumor cells, including upregulation of immunoproteasome subunit gene expression. The number of presented peptides significantly increased upon IFNγ treatment, but neither binding affinity nor processing signatures were altered. Addressing immunogenicity, she showed that only about 10% of predicted mutations in multiple mouse tumor models were immunogenic in vivo and discussed how the position in the peptide of the mutation, its context for anchor mutations, and whether it bears similarity to immunogenic microbial peptides can be taken into account.
Following Delamarre, Nina Bhardwaj discussed her team’s use of OpenVax, a toolbox of open source software for mutation calling and peptide:MHC binding prediction to identify highly-immunogenic neoantigens. Utilizing this software, they analyzed the B16F10 mouse melanoma cell line and tested immunogenicity of 15 of the variants using a peptide and adjuvant nanoparticle co-delivery system developed by Avidea Technologies. About half of the predicted epitopes were immunogenic, and control of B16F10 correlated with the affinity, stability, and magnitude of CD8+ T cell responses. They then used their software to predict patient-specific neoantigens and conducted a personalized neoantigen vaccine trial utilizing long peptides and the double-stranded RNA adjuvant pICLC. Polyfunctional CD4+ and CD8+ T cell responses were observed. Finally, as a possible off-the-shelf vaccine strategy, they identified a common set of genetic mutations between various myeloproliferative neoplasms and validated a number of immunogenic epitopes using in vitro and ex vivo assays.
Closing out the neoantigen vaccine session, Catherine Wu summarized her team’s research published in 2017, in which personalized neoantigen vaccines exhibited immunogenicity and clinical efficacy in high-risk melanoma patients. She then introduced results from a recent clinical trial that employed the same neoantigen vaccination strategy against glioblastoma (a cancer with a much lower mutation burden) following surgical resection and standard-of-care radiation therapy. Conveniently, surgical resection requires about eight weeks of recovery and radiotherapy, allowing Wu’s group ample time to analyze the tumor and develop personalized vaccines. Eight patients were dosed. Six of the patients had received the corticosteroid dexamethasone as part of their standard care and did not respond immunologically to the vaccine. However, the two patients who had not received corticosteroid treatment showed strong immunological responses. Unfortunately, disease recurred in all the patients and none of the enrolled patients in this study survived; however, this did allow access to post-therapy tumor samples, including one fresh tumor sample from a responding patient. Using single cell RNA sequencing and targeted TCR sequencing, they identified multiple T cells common to the peripheral blood and the brain tumor and were ultimately able to determine that a vaccine-induced neoantigen-specific CD4+ T cell had infiltrated the tumor.
In a later session, focusing on the peptide epitope, Michal Bassani-Sternberg described her team’s multi-disciplinary approach to discovering and validating potential peptide epitopes. Multiple features (peptide binding strength and stability, processing, expression, TCR recognition, clonality, etc.) impact presentation and immunogenicity of neoantigen peptides and must be considered when choosing which among the possible thousands of native and neoantigen peptides to target for an immunotherapy product. Mass spectrometry (MS), genomics and transcriptomics, and T cell functional assays (in vitro and ex vivo) are the cornerstones of Bassani-Sternberg’s approach. MS has been used both to detect native peptide epitopes, which vary between patients and in different cell lines within a patient, and to improve algorithmic prediction rules. Analysis of their MS databases suggested that proteasomal spliced epitopes represent at most only 2-4% of the peptide ligandome. Detecting patient-specific neoantigens with MS has been more challenging, as MS likely favors detection of only the most abundant neoantigens. Bassani-Sternberg, however, described the case of a patient for whom 8 different neoantigen epitopes were detected by MS, and two of these were likely immunogenic based on analysis of patient peripheral blood mononuclear cells (PBMCs).
In a separate session, attempting to answer the question of why the response to anti-PD-1 therapy is so low in patients with epithelial ovarian cancer (EOC), George Coukos dug deeply into the pre-existing and vaccine-induced immune responses in ovarian cancer. His team found that tumor-infiltrating lymphocytes (TILs) in EOC are specific for a number of tumor-associated antigens, located in the tumor and not the stroma, are PD-1 positive with an activated, rather than exhausted, phenotype, and respond to anti-PD-1 blockade. Since none of these observations explained the low therapy response rate, Coukos and his team checked for neoepitope-specific recognition by the CD8+ TILs, which they also found. Unable to explain the lack of response to PD-1 blockade based on detailed T cell analysis, Coukos called for a broad “systems immunology” approach, integrating multiple disciplines and approaches to solve such multi-dimensional problems. Coukos closed by describing a separate study in which his team explored using autologous dendritic cells (DCs) pulsed with autologous whole-tumor cell lysate as a personalized vaccine, administered either alone or in combination with immunomodulatory drugs that were meant to normalize the endothelium (anti-VEGF) and reduce Tregs (cyclophosphamide). The vaccine, which was well-tolerated, induced new and pre-existing antitumor T cell responses, which correlated with better outcome.
In a session on overcoming treatment resistance, Weiyi Peng discussed how combining an OX40 agonist with PI3Kβ selective inhibitor (GSK2636771) enhances T cell immunity. In tumors with loss of tumor suppressor PTEN, the PI3K pathway is activated, leading to an immunosuppressive TME with reduced trafficking of T cells to the tumor and diminished susceptibility to T-cell mediated apoptosis. A costimulating OX40 agonist synergizes with a PI3Kβ inhibitor to delay progression of PTEN-loss tumors and prolong survival of mice with melanoma by enhancing the activation of CD8+ TILs, increasing the serum levels of cytokines produced by memory/effector T cells, and promoting the proliferation of immune cells involved in antitumor activity.
Mohammed Milhem discussed the results of a phase Ib clinical trial in 69 patients evaluating intratumoral CMP-001 (a TLR9 agonist designed to activate tumor-associated plasmacytoid DCs and induce IFN production and antitumor CD8+ T cell responses) in combination with systemic pembrolizumab in patients with metastatic melanoma resistant to PD-1 checkpoint inhibition. In addition to meeting its primary endpoint of safety, Milhem’s team saw an overall response rate of 22 percent, with a trend toward higher overall response at particular doses and schedule. Notably, patients who responded to therapy saw a rapid reduction in the size of both injected and non-injected tumors, indicating a systemic immune response, and a transcriptional signature reflective of an inflamed tumor.
CD40 signaling is an emerging player in the field of cancer immunotherapy due to its role in determining whether cytotoxic T lymphocytes become primed or tolerized. Hayley Ma presented research which showed that in an immunocompetent mouse model of pancreatic ductal adenocarcinoma (PDA), a cancer with low T cell infiltration at baseline, the addition of agonistic CD40 to GVAX and anti-PD-1 resulted in 100% tumor clearance. The triple combination therapy led to changes in monocytic and DC infiltration and maturation, and a decrease in granulocytic myeloid-derived suppressor cells (G-MDSCs) that correlated with improved response. According to Ma, changes in the myeloid populations likely translate to an increase in infiltrating CD8+ T cells, which ultimately mediated tumor clearance.
Alex Morrison showed that in mouse models of PDA, a combination of agonistic CD40 monoclonal antibody and immune checkpoint blockade (anti-PD-1 and anti-CTLA-4 monoclonal antibodies) led to tumor regression in 100% (7/7) of mice and some apparent cures. Early tumor regression was primarily dependent on CD4+ T cells, while long-term remission was dependent on both CD4+ and CD8+ T cells. Further, these studies showed that agonistic CD40 and immune checkpoint blockade play non-redundant roles in generating a strong antitumor T cell response.
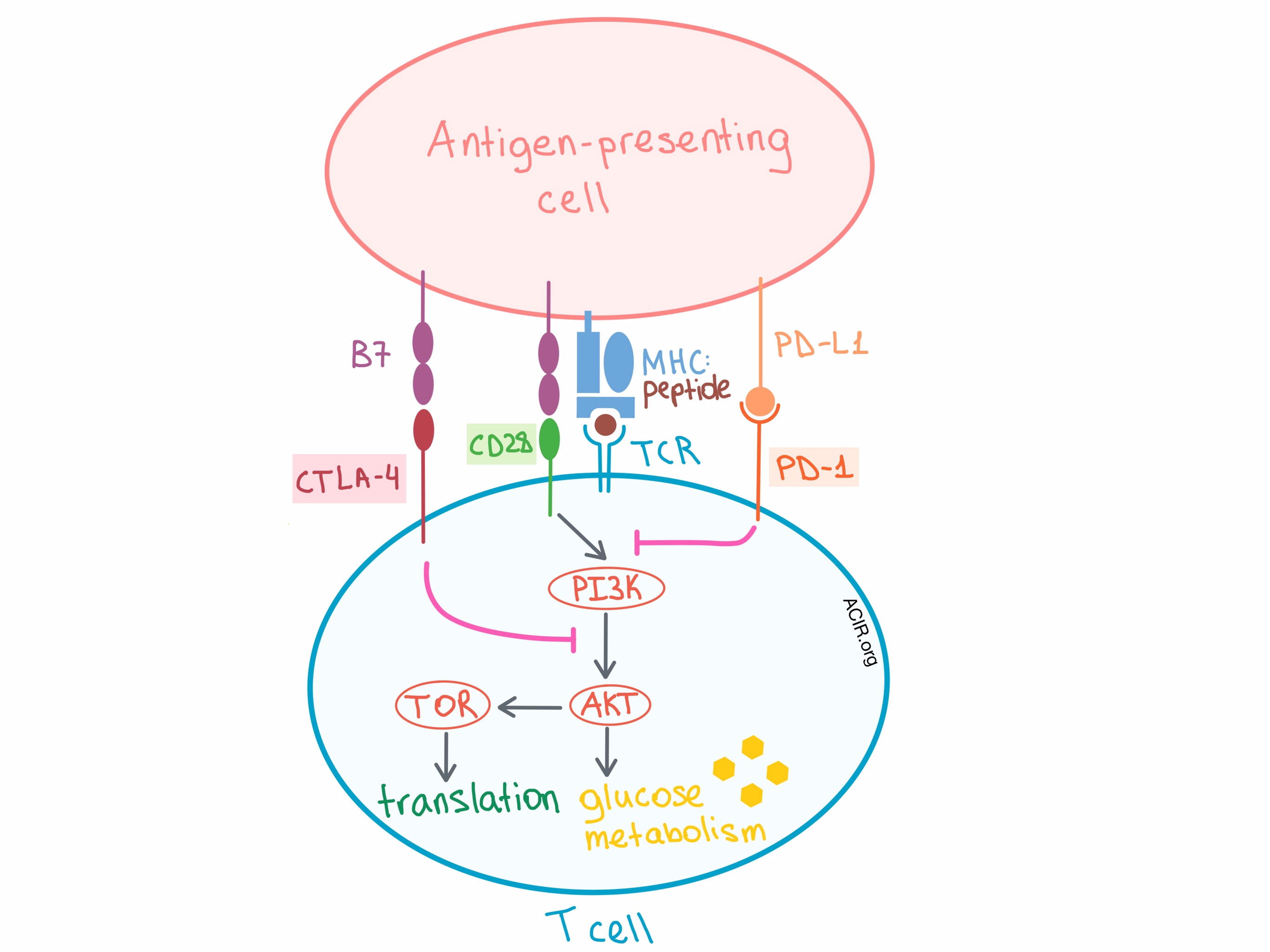
In a session on cancer metabolism, Craig Thompson discussed the fight for nutrients between immune cells and cancer cells that occurs within the tumor microenvironment (TME). Both T cells and tumor cells require two signals to proliferate. In T cells, signal 1 occurs via the TCR, and signal 2 is via the costimulatory CD28 molecule. While signal 1 modifies specific gene expression, signal 2 amplifies that expression. Specifically, CD28 costimulation is required to upregulate/maximally stimulate glucose transporter expression and glycolysis, and this occurs via PI3K/AKT pathway activation. On the other hand, upregulation of CTLA-4 and PD-1 in recently activated T cells leads to mitigation of this response. Both CTLA-4 and PD-1 signaling block CD28-dependent AKT activation, but by independent mechanisms, leading to suppression of glucose uptake at the glucose metabolism stage of the pathway. In over 70% of cancers the PI3K/AKT/TOR pathway, which fuels cell growth, is mutated.
So, is metabolism an explanation for why checkpoint blockade only works in a fraction of patients and tumor types? Metabolically, when T cells are chronically stimulated and become exhausted within the TME, there is induction of HIF-1α and persistent aerobic glycolysis. Under such conditions, oxygen consumption goes down, extracellular acidification rate goes up, T cells are depleted of substrates for chromatin modification reactions, reactive oxygen species (ROS) are induced, and, importantly, mitochondria do not have the energy to sustain cell growth and proliferation. Thompson’s group found that adding N-acetylcysteine to chronically stimulated T cells reduces ROS stress and rescues the T cells from exhaustion. In a separate experiment, they found that the most commonly depleted nutrient in human tumor tissues is not glucose, but non-essential amino acids (AAs), especially glutamine. Reducing, but not depleting, the AAs to 5% of normal levels can lead to immunosuppression because under these conditions, CD4+ T cells transform from an exhausted phenotype to an immunosuppressive Treg phenotype!
Susan Kaech continued the metabolic discussion in her talk on antitumor T cell metabolism within the TME. Her research attempted to answer the question of how metabolic adaptation by the T cells in the TME contributes to their dysfunction. Hyper-glycolytic tumors evade the immune system more successfully. Within such tumors, CD4+ T cells lose their functionality and produce less IFNγ. In addition, there is little T cell infiltration into the hypoxic areas of the tumors. Under such conditions, T cells in the tumor were found to increase fatty acid (FA) uptake compared with T cells in the spleen via CD36, which increases FA uptake and regulates FA homeostasis in CD8+ T cells. The metabolism of certain FAs, such as oleic acid and linoleic acid, suppresses T cell effector function, thus promoting tumor growth. Although the exact mechanism of T cell impairment is yet to be determined, CD36 can now be considered a new checkpoint.
In a session on genomic approaches in cancer immunology, Nir Hacohen discussed the targets, drivers, and resistors of the human antitumor immune response, focusing on tumor antigens, genetic and epigenetic changes, and the types and states of immune cells within the TME. System immunology approaches illuminated the co-evolution of tumor and immune response over time, showing that some clones are eliminated while others grow. This pruning of neoantigens in human tumors involves T cells in an HLA-dependent manner. In addition, genetic and epigenetic factors lead to B2M deficiency, which may impact survival. In a study of 23 melanoma patients treated with anti-PD-1 (sometimes in combination with anti-CTLA-4) with known clinical outcomes, the researchers searched for predictive markers and potential new targets by extensive single cell RNA-sequencing and subsequent bioinformatic clustering. They found that the pre-therapy ratio of memory/exhausted CD8+ T cells strongly correlated with response. This ratio combined with B2M mutation status and HLA status was a nearly perfect predictor of response.
Exploring the tumor myeloid microenvironment, Stefani Spranger described the relationship between Batf3-DCs and antitumor T-cell response. Conventional DCs that express CD103 and the transcriptional factor Batf3 are rare but necessary for the presentation of tumor antigens to CD8+ T cells in the tumor-draining lymph nodes. The researchers found that tumors lacking such DCs do not generate an antitumor immune response and are not responsive to immune checkpoint blockade. This is due not only to a lack of T cell priming but also to deficient T cell infiltration into the tumor, as tumor control in Batf3-DC-negative tumors does not improve even with adoptive T cell transfer. Spranger and her team found that tumor-residing Batf-3 DCs are the predominant source of CXCR3 chemokine ligands (CXCL9 and CXCL10), and are required for the recruitment of effector T cells, demonstrating another contribution of tumor-residing DCs to the local antitumor response.
Benjamin Youngblood discussed the epigenetic regulation of T-cell exhaustion and its implications for cancer immunotherapy. The researchers utilized mouse tumor and chronic viral infection models to explore the role of de novo epigenetic programming in establishing an exhausted T cell phenotype that is resistant to immune checkpoint blockade. They found that DNA methyltransferase Dnmt3a promoted de novo DNA methylation programming over time in effector CD8+ T cells during chronic exposure to antigen, leading to their exhaustion. This reprogramming restricted T cell expansion during PD-1 blockade therapy, while knocking out Dnmt3a led to a significant expansion following anti-PD-1 therapy. Although the expanded Dnmt3a- T cells are PD-1hi, they are not functionally exhausted due to their inability to reprogram the T cell genome. Overall, these results point to epigenetic reprogramming of T cells as a potential approach to enhancing T cell-based cancer therapy response.
Building on earlier work targeting highly expressed CD25 as a mechanism for Treg cell depletion, Sergio Quezada showed that while currently used CD25 targeting antibodies have limited efficacy due to the fact that they effectively prevent critical IL-2 signaling to effector T cells, an improved depleting antibody, αCD25NIB, did not block IL-2 binding and was more powerful as a monotherapy and in conjunction with checkpoint blockade. In the immunogenic CT26 model, a single dose of αCD25NIB prevented tumor growth in an IL-2 dependent manner; in the “cold” B16F10 model, a single dose of αCD25NIB along with GVAX vaccine provided a substantial improvement in tumor control over GVAX alone.
To begin to better understand why targeting of the WT1 protein with TCR-engineered T cell transfer was effective prior to, but not following AML relapse, Kelly Paulson analyzed cells from a single patient and demonstrated that tumor escape was not mediated by downregulation of WT1 or HLA, but by downregulation of a single subunit of the immunoproteasome, β1i, which led to defective antigen processing of the WT1 epitope. To avoid this type of immunoediting, she suggests taking aim at multiple immune targets and improving early detection and treatment options.
On the topic of recent advances in CAR T cell therapy, Renier Brentjens discussed his team’s development of cytokine-producing “armored” CAR T cells, which secrete IL-12 to induce Th1 CD4+ T cell response and enhance IFNγ production. In mouse models, IL-12-secreting armored CAR T cells were able to eradicate advanced ovarian tumors, increase T cell proliferation, central memory populations, and survival, and were resistant to Treg-mediated inhibition. Brentjens’s team also developed IL-18-secreting armored CAR T cells to induce a proinflammatory response, which enhanced long-term survival in mice. These T cells showed enhanced proliferation and in vivo persistence and led to increases in IL-18, IFNγ, and TNFα, without increasing IL-6, which is associated with cytokine release syndrome. Another armored CAR T cell design included utilizing CD40L which helped to mature DCs, enhance T cell proliferation and cytokine secretion, and improve survival.
Finally, Toni Ribas was this year’s awardee for the AACR-CRI Lloyd J. Old Award in Cancer Immunotherapy to celebrate outstanding achievements in the field of cancer immunotherapy in honor of the father of modern cancer immunotherapy. Dr. Ribas has been an outstanding physician-investigator at the UCLA Medical Center, contributing significantly to the fields of adoptive T cell therapy, checkpoint therapy, and the mechanisms of tumor resistance to immunotherapy. Based on the theme that “the T cell is the drug,” he walked through old and new work on improving the functionality of adoptive T cells through genetic engineering. Among the new concepts described were the genetic programming of hematopoietic stem cells as a source of TCR transgenic cells, the use of modified versions of IL-2 that target the adoptive cells more effectively, and the application of non-viral CRISPR/CAS9 gene editing to precisely genetically target and accelerate the production of future T cell therapies.
by Lauren Hitchings, Anna Scherer, and Ed Fritsch



