
Last week, the ACIR team attended the AACR Annual Meeting 2022 in New Orleans, LA. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
T cell therapies
Robbie Majzner
John Haanen
Marcela Maus
Noam Levin
Angela Aznar
Cancer vaccination strategies and vaccine-elicited antibodies
Leila Delamarre
Michelle McKeague
T cell biology
Ton Schumacher
Samir Khleif
Tumor immune environment
Juan Dubort
David Bellovin
Casey Ager
Salah-Eddine Bentebibel
Ira Mellman
Miriam Merad
Lloyd J. Old Award in Cancer Immunology
Ira Mellman
T cell therapies
Major tumor regressions in H3K27M-mutated diffuse midline glioma (DMG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T cells - Robbie Majzner - Stanford University.
Earlier this year, Robbie Majzner and colleagues reported the very early results from a dose-escalating phase I clinical trial, in which 4 pediatric patients with diffuse intrinsic pontine glioma (DIPG) or other H3K27M-mutated diffuse midline gliomas (DMGs) were treated with CAR T cells targeting GD2. In this AACR presentation, Majzner reported on a total of 11 patients treated at two dose levels. For each patient, the first dose was given intravenously (IV) (with lymphodepletion) while subsequent doses were administered intracerebroventricularly (ICV) (without lymphodepletion) via an Ommaya reservoir, which was also required for monitoring intracranial pressure and removing cerebrospinal fluid to alleviate tumor inflammation-associated neurotoxicity (TIAN) – a common, but expected and manageable adverse event (AE) associated with this treatment. For IV administration, patients at the first dose level experienced no dose-limiting toxicities, while several patients at the second dose level experienced high-grade cytokine release syndrome (CRS), pointing to the first dose level as the maximum tolerated dose for IV administration. However, the higher dose level was well tolerated for ICV administration. Early results showed that ICV administration induced stronger cytokine profiles in the cerebrospinal fluid and weaker profiles in the serum, consistent with more targeted treatment and less severe AEs. Out of ten evaluable patients, nine experienced radiographic and/or clinical benefit after IV infusion, and went on to receive a median of 4 subsequent ICV infusions of GD2-CART. Among all 11 patients, one achieved a complete response, two achieved partial responses, and 6 achieved stable disease (RANO criteria). Patients lived for a median of 20 and 18 months from diagnosis at dose levels 1 and 2, respectively, though it is worth noting that patients enrolled later in the trial were treated much earlier after diagnosis compared to those who had enrolled at the first dose level. Four patients continue to receive ICV infusions on study and have experienced continued clinical and radiographic benefits. Highlighting results from individual patients, Majzner noted a 17-year-old male with DIPG who experienced a complete response that was durable for 10+ months, and was accompanied by improvements in walking, sensation, and hearing. Another patient, a 4-year-old female with DIPG, had slight radiographic benefits for 12+ months, but experienced significant clinical improvements. She has gone from not being able to walk or smile, to running, climbing, and smiling wide. A 5-year-old girl has shown improvements in right-sided spasticity and weakness, increased weight and height, better balance and coordination, and reduced drooling and hypophonia. Showing a series of progressively improving coloring pages completed by a 4-year-old patient before and on treatment, Majzner noted the importance of evaluating patient-reported outcomes that go beyond the scope of scans and lab values and show how novel therapies like these are improving lives. These patient-reported outcomes will play a more significant role in clinical evaluation as Majzner and colleagues move forward with the trial. They will also be testing a new clinical trial arm in which treatment will only be administered ICV without lymphodepletion.
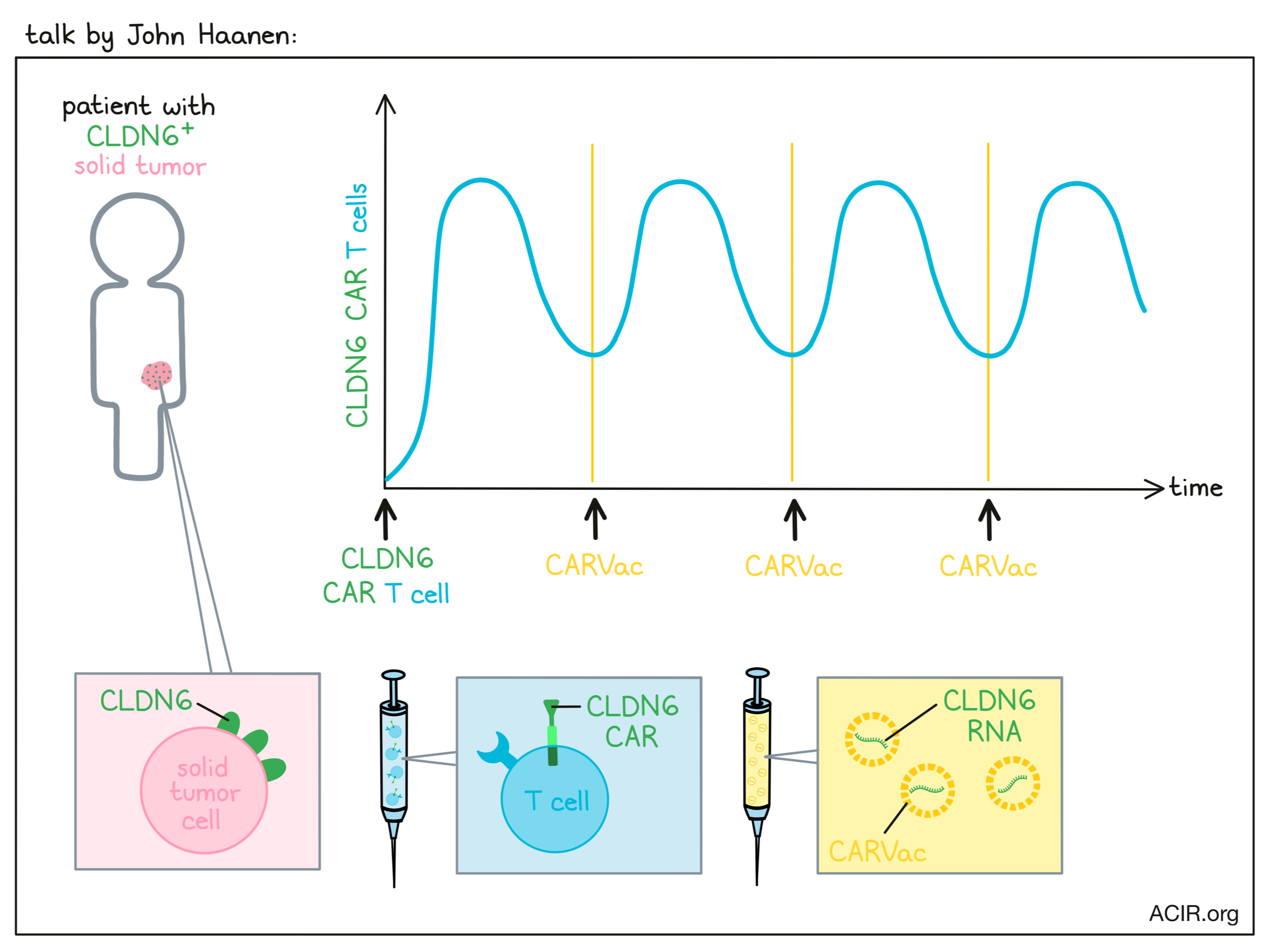
BNT211: A Phase I trial to evaluate safety and efficacy of CLDN6 CAR-T cells and CARVac-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumors - John Haanen - Netherland Cancer Institute (NKI).
Speaker John Haanen discussed a recent clinical trial that evaluated the safety and efficacy of a novel two-part strategy for targeting solid tumors with CAR T cells specific for Claudin-6 (CLDN6) – a molecule expressed during development and re-expressed in a number of cancers. Patients in this trial were heavily pre-treated and had a variety of solid tumor types, including testicular and ovarian cancer. In addition to the CAR T cells, some cohorts were also treated with CARVac – an RNA vaccine encoding CLDN6 that is used to mediate periodic in vivo expansion of CLDN6-CAR T cells by inducing CLDN6 expression and presentation in antigen-presenting cells. Looking first at the dosing of CAR T cells alone, the researchers observed some dose-limiting toxicities at their second dose level. Out of 16 treated patients, most experienced adverse events, particularly cytokine release syndrome (CRS; accompanied by elevated levels of IL-6), elevated lipase (with no apparent toxic effects), and events associated with pre-treatment lymphodepletion. Thus, the first dose level was determined to be the maximum-tolerated dose. Prolonged cytopenia was noted in patients who had recently undergone high-dose chemotherapy, and a new cohort was opened in which such patients would undergo reduced lymphodepletion to prevent this. Among 14 evaluable patients across all cohorts (dose levels 1 and 2 with and without CARVac), the overall response rate (ORR) was 43% and the disease control rate (DCR) was 86%. Patients who initially showed partial responses showed evidence of deepening responses over time, and one patient achieved a complete remission. Despite the toxicity observed at the second dose level, treatment at this dose was more effective. Patients being treated at dose level 2 plus CARVac had a 75% ORR and a 100% DCR. At both dose levels, the addition of CARVac contributed to improved responses. In at least some patients, CARVac was shown to support the initial engraftment of CAR T cells and induce re-expansion of CAR T cells with each administration. Interestingly, CARVac also appeared to upregulate cytokine receptors, including the IL-2 and IL-7 receptors, on CD4+ CAR T cells, which could enhance T cell proliferation, activation, survival, and homeostasis. In the one patient who achieved complete remission (this patient did not experience CRS), the researchers observed that CAR T cells showed a dominant EMRA phenotype and persisted for over 150 days. While there are still a number of challenges to come in using CAR T cells to treat solid tumors, this trial, thus far, has shown promising results against difficult disease targets.
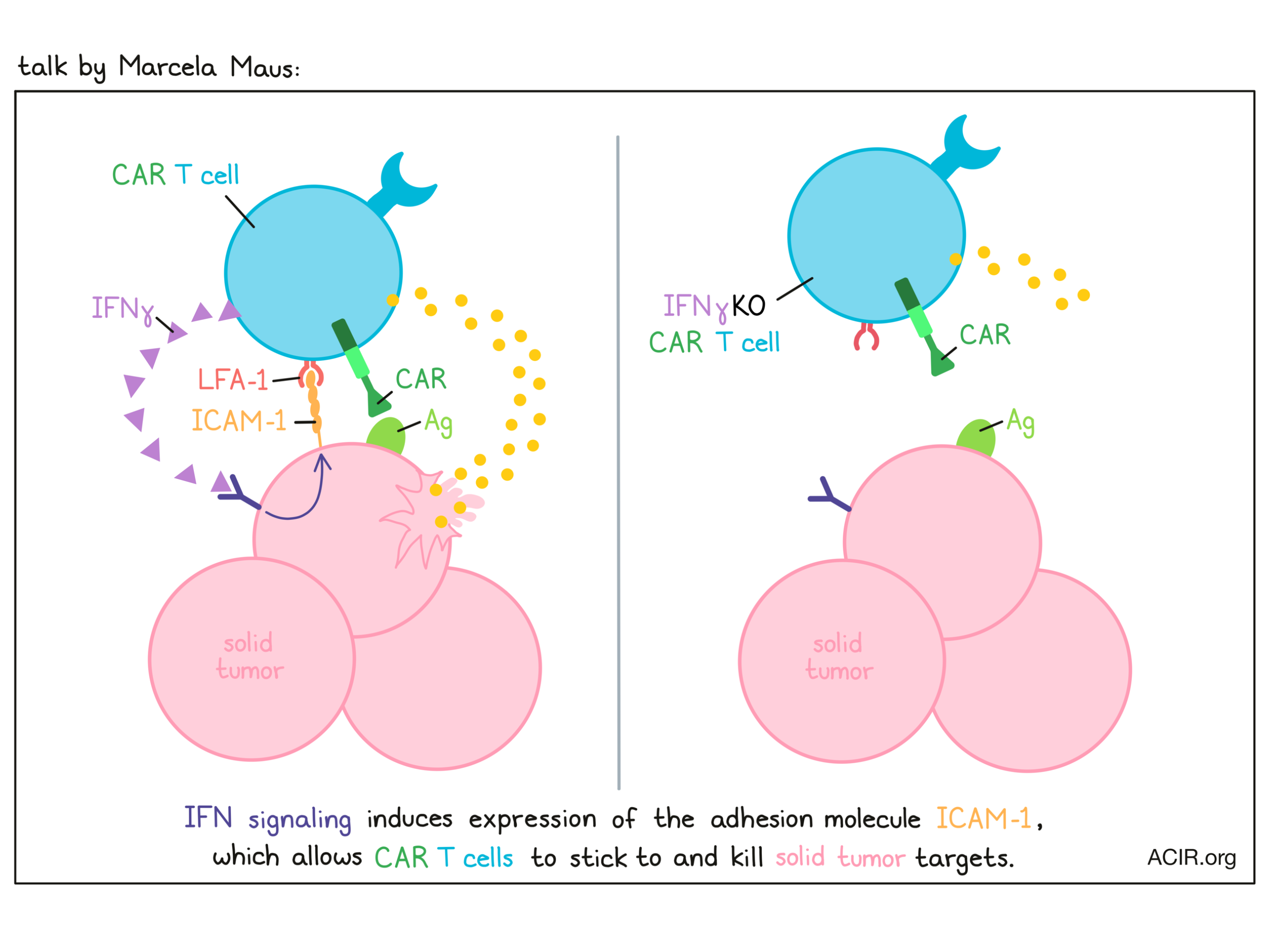
Mechanisms behind CAR T cells - Marcela Maus - Massachusetts General Hospital (MGH).
In recent efforts to reduce side effects associated with CAR T cells, Marcela Maus and colleagues uncovered an interesting pattern in which the production of IFNγ by CAR T cells was not required for the killing of liquid tumors, but was required for the killing of solid tumors. Digging deeper into the mechanism underlying this phenomenon, Maus’s team performed a genome-wide CRISPR screen in murine glioblastoma (GBM) models to determine which gene or genes conferred resistance to CAR T cells. Expectedly, EGFR was the top hit, but interestingly, other top hits included IFNγ receptor pathway genes, including JAK2, IFNGR1, and IFNGR2. The importance of this pathway was confirmed by generating IFNγR1 KO GBM, which was indeed more resistant to CAR T cell-mediated killing than wild-type GBM, both in vitro and in vivo; this phenomenon was also observed across other solid tumor models with different antigens and histologies. Looking at IFNγR1 KO in liquid tumors, the effects observed were nearly the opposite, as these cells could still be as readily killed by CAR T cells as wild-type tumor cells. To better understand this, the researchers investigated the transcriptional effects of CAR T cells on tumor cells, and vice versa, after exposure to one other. CAR T cells exposed to IFNγR1 KO tumors were transcriptionally similar to those exposed to WT tumor cells, while IFNγR1 KO solid (but not liquid) tumor cells exposed to CAR T cells were quite different. The IFNγR1 KO solid tumor cells had lower adhesion and migration scores in a novel cell-binding assay compared to WT cells, along with lower binding avidity to CAR T cells. Turning their attention towards expression of adhesion molecules, the researchers found that while wild-type tumor cells upregulated ICAM-1 after coculture with CAR T cells, IFNγR1 KO tumor cells did not. Testing the functional relevance of this observation, the researchers found that blockade of ICAM-1 or its binding partner LFA-1 in wild-type cells recapitulated the effects of IFNγR1 KO. Further, forced overexpression of ICAM-1 in IFNγR1 KO tumor cells restored their binding avidity and sensitivity to CAR T cell-mediated killing. Overall, these results showed that while IFNγ may not play a direct role in killing target tumor cells, engagement of the IFNγR pathway helps CAR T cells to stick to and and ultimately kill solid tumor targets.
Identification of T-cell receptors targeting RAS hotspot mutations using TIL IVS in human cancers for use in cell-based immunotherapy - Noam Levin - National Cancer Institute (NCI).
Given the high prevalence of KRAS hotspot mutations across diverse cancer types, identification of effective KRAS mutation/HLA allele-specific TCRs would potentially be a valuable therapeutic strategy. Building on the initial encouraging clinical result of a patient with multiple metastases treated with a TIL product highly enriched in polyclonal KRAS G12D-specific T cells (HLA Cw*08:02), Noam Levin compared two earlier procedures to obtain additional KRAS-specific TCRs (standard TIL fragment culture [TIL] or in vitro stimulation of patient PBMCs [PBL IVS] followed by identification of specific T cells through antigen stimulation and 4-1BB/OX40 upregulation readout) with a new procedure comprising in vitro stimulation and TIL culture [TIL IVS] followed by specific T cell identification. Although the older procedures did identify multiple different TCRs restricted by different HLA alleles, there was little overlap between TCRs from tumor and peripheral blood from the same patient, suggesting limited sensitivity. The new TIL IVS procedure was used on 25 patients. All of these patients also underwent a standard TIL screen, and 6 of these were screened by PBL IVS, allowing direct comparisons of techniques. 8 standard TIL and 2 PBL IVS patients yielded TCRs, while all 8 + 2 of these and an additional 15 patients were positive by TIL IVS. The numbers of individual TCR clonotypes similarly increased from 13 + 2 to 46. To confirm that the more comprehensive recovery of TCRs maintained good properties, 2 TIL-derived and 2 TIL IVS-derived TCRs (all KRAS G12V mutation and HLA Cw*01 restricted) were compared. Functional avidities, tumor recognition in vitro, and tumor killing in immune-deficient mice were all comparable. The library of available KRAS TCRs isolated by these procedures now includes 48 TCRs recognizing 4 different KRAS mutations and restricted by multiple HLA class I and class II alleles, covering 45-60% of all patients expressing a KRAS mutation.
Mechanisms of CAR T cell dysfunction and identification of transcription factors that drive the exhaustion phenotype - Angela Aznar - University of Pennsylvania.
To improve effector CAR T cell function in solid tumors, Angela Aznar assessed mechanisms of CAR T dysfunction and transcription factors involved in this phenotype. The researchers first developed an in vitro model of chronic antigen exposure (CAE) to drive CAR T cell dysfunction. The model was created using anti-mesothelin (MSLN.bbz) CAR T cells (M5) that were cocultured with MSLN-expressing AsPC-1 pancreatic cancer cells at a low effector-to-target ratio. After chronic exposure to antigen, scRNAseq was conducted on the CAE CAR T cells to define dysfunction signature genes upregulated after CAE. NK markers and checkpoint expression on these cells revealed that CAR dysfunction was associated with a CD8 T-to-NK-like T cell transition (CD3+, KLRB1+, KLRC1+). M5 CAR T cells in vivo in therapy-resistant recurrent tumors also presented with this signature, while MSLN was still expressed on the tumor. In another context, anti-NY-ESO-1 TCR T cells in tumor-infiltrating lymphocytes of mice in which tumors were not completely cleared were also found to be high in these dysfunction signature genes. To assess what the main drivers of these signatures were, an upstream regulator analysis was performed based on bulk and single-cell RNAseq data, revealing ID3 and SOX4 among the differentially expressed transcription factors found uniquely in the dysfunctional T cell clusters. When ID3 or SOX4 was knocked out of the CAR T cells, the in vitro effector function improved in the CAE assay and the dysfunction scores in these cells decreased. Finally, in an AsPC-1 xenograft model, it was shown that these CAR T cells with knockouts showed enhanced antitumor efficiency, pointing to possible ways to improve CAR T cell function for solid tumors.
Cancer vaccination strategies and vaccine-elicited antibodies
Beyond tumor antigen immunogenicity: Determinants of protective immunity induced by therapeutic cancer vaccines - Leila Delamarre - Genentech.
With an aim to improve the success rate for selection of neoantigens for personal mutation-based vaccines, Lelia Delamarre and team focused on peptide presentation, a critical gateway to immunogenicity and efficacy. For presentation, suspected critical features (such as amino acid sequence, flanking regions, and likely MHC contact residues) of MHC-eluted peptide ligands from 15 peripheral blood sources and 124 human cancer cell lines spanning 123 MHC alleles were used to instruct machine learning approaches. The resulting algorithm significantly outperformed the current benchmark, netMHCpan v4.0, in terms of antigen presentation, as well as better predicted epitopes that were shown to be recognized by T cells, and hence were immunogenic. To explore immunogenicity and efficacy, Delamarre began by summarizing previously published analyses of immunogenicity of predicted neoantigens in four mouse models. Only about 10% of predicted, expressed neoantigens across these models were immunogenic, as adjuvanted long-peptide vaccines and the key features correlating with immunogenic epitopes were high absolute affinity (for non-anchor residue mutations) and high relative mutated/wild type affinity (for anchor residue changes). Interestingly, parameters such as hydrophobicity, bulkiness, and similarity to wild-type epitopes or to microbial antigens were not correlative, contrasting with prior published studies. Ultimately, efficacy is the key readout, which requires features beyond immunogenicity. such as persistence, trafficking, and on-target cytolysis. To address this, two different sets of ten predicted and known immunogenic epitopes from MC38 tumors (Deca1 and Deca2) were prepared as a polyepitopic construct and delivered to mice via the clinically used RNA-LPX format, which provides both enhanced DC uptake and self-adjuvanticity (TLR7/8 stimulation). Although both vaccines were comparable in terms of overall immunogenic response and phenotype (revealed by single-cell RNA sequencing) in non-tumor-bearing animals and in terms of prophylactic tumor control, control of established tumors by 3 weekly vaccinations was dramatically different; only Deca2 controlled tumor growth. This effect was not due to differences in overall or tetramer-positive CD8+ T cell infiltration into tumors between the two vaccines. However, analysis of the phenotype of neoepitope-specific tumor-infiltrating T cells in vaccinated tumor-bearing animals was different from those in vaccinated tumor-free animals for both constructs. Importantly, the phenotype of clonal, neoepitope-specific T cells in spleens was further altered upon infiltration into the tumor for Deca2, but not for Deca1. Deca2 intratumoral T cells were more active and expressed higher levels of granzyme B and perforin. It is the focus of ongoing investigation to learn what signals differentially modulate the intratumoral T cell phenotype (TCR avidity, tumor epitope presentation, level of intratumoral DC cross-presentation).
Patient-derived, vaccine-elicited, anti-MUC1 antibodies directly target tumor cells for elimination via multiple immune mechanisms - Michelle McKeague - University of Pittsburgh.
Mucin-1 (MUC1) is a cancer-associated antigen expressed in healthy tissue, but overexpressed in cancer. The extracellular domain contains a variable number of multiple tandem repeats (VNTR) of a heavily glycosylated domain, which are hypoglycosylated in multiple cancer histologies, exposing the peptide backbone as a novel B and T cell epitope. Michelle McKeague presented data on antibodies obtained from individuals with a high risk of colon cancer with polyps or adenomas, who were treated with a non-glycosylated MUC1-100mer peptide vaccine (containing 5 repeats of the normally glycosylated domain) and pICLC (Hiltonol®). Cloned antibodies from these individuals only bound tumor tissue, not healthy tissue expressing MUC1. In an attempt to assess the immune effector mechanisms of these antibodies, McKeague used various in vitro and in vivo assays assessing anti-cancer activity. In vitro coculture assays showed that the antibodies induced tumor cell killing via cytokine release (ADCR), NK cell killing (ADCC), monocyte phagocytosis (ADCP), and neutrophil trogocytosis (ADCT), but not complement cytolysis (CDC). When testing the H15K6 antibody, which binds to the peptide repeat region of the MUC1 VNTR, using the human MUC1-expressing Ad755 murine breast cancer cell line in vitro, there was no evidence of ADCC or CDC, but there was ADCT and ADCP. Treating Rag-/-γc-/- mice bearing this breast cancer tumor with the human antibodies resulted in delayed tumor growth and increased survival. Having this pool of human antibodies directed against the same antigen provides a unique system to address various characteristics of antibody binding, such as antibody affinity, epitope quantity, epitope proximity to the membrane, and membrane association. To study these characteristics, various structural forms of the MUC1 protein were transduced into Jurkat cells and used to perform functional assays with respect to the amount of bound antibody. This revealed that the amount of antibody binding did not perfectly correlate with the ability to mediate ADCC and ADCP, suggesting other properties of antigen–antibody interactions are contributing to their efficacy. ADCT did correlate with the amount of antibody binding. Underway are studies to test the antibodies in tumor-bearing, immunocompetent mice bearing a human FcR transgene.
T cell biology
T Cell Reactivity in Human Cancer - Ton Schumacher - Netherland Cancer Institute (NKI).
With the theme of taking an engineering approach to dissect the activities of immune cells in immune oncology therapies, Ton Schumacher described novel technologies specifically designed to address two important outstanding biological questions: (1) Do intratumoral T cells respond to PD-1 blockade? and (2) What is the developmental origin of intratumoral stem-like T cells? A large bank of patient-derived tumor fragments (PDTF) were prepared from tumors of multiple histologies. Each resected tumor was fragmented into multiple “tumor avatars” and frozen. PDTFs provide an opportunity to understand baseline tumor-immune functional characteristics, as well as recapitulate therapeutic treatment by ex vivo controlled perturbations (for example, treatment with anti-PD-1 antibody); multiple replicates provide the opportunity to investigate intratumoral and interpatient heterogeneity. Building on earlier published work, markers of cell activation and cytokine secretion enhanced by anti-PD-1 clustered ex vivo samples from responding and non-responding patients, and a response score based on the most contributory parameters revealed an association between avatar response and observed clinical response. Interestingly, analysis of individual fragment responses from the same patient showed both significant heterogeneity in terms of response or non-response by individual fragments, and consistency of response pattern across responding fragments within a patient, but different response patterns between patients, pointing to an important need to further study heterogeneity. Turning to evaluating T cell phenotypes, a larger patient data set (226 PDTFs) resolved 4 categories of anti-PD-1 responders, independent of tumor type: IFNγ response, TH1, CXCL11 with low cytotoxicity, and IL-2/6 with high cytotoxicity. This adds key functional distinctions that can be added to the cell characterization atlas. Following treatment with anti-PD-1, interestingly, CD39+ cells, typically characterized as late exhausted cells, were activated (as measured by CD137 and granzyme B expression), indicating that such cells may play some role in the tumor microenvironment following anti-PD-1 therapy. Single-cell RNA sequencing revealed no major transcriptional changes in such cells, suggesting that translational control, but no lasting re-wiring was occurring. Together with data from other labs, this result is consistent with a model in which such pre-existing exhausted cells are activated by anti-PD-1 treatment to produce IFNγ and chemokines, triggering infiltration of new cells, including stem cells that are clearly important to durable responses. Finally, to understand the origin of such stem-like cells, a “Division Recorder” system was built, which monitored the number of cell divisions an individual cell experienced over a long period of time in vivo. Following in vivo transfer of such marker cells and infection challenge, coupling of single-cell RNA sequencing and the division recorder revealed two subsets of central memory cells, a more replicated subset with “effector” signature and with low proliferative potential, and a quiescent “multipotent” subset that replicated less, but with high proliferative potential. Multipotent cells also had a core stem cell memory gene expression signature, consistent with self-renewal. These data fit a linear model of differentiation of T cells from antigen activation to effector cells.
T cell priming, a crucial strategy for reversal of immunotherapy resistance - Samir Khleif - Georgetown University.
Given that many patients do not respond to anti-PD-1 immune checkpoint blockade (ICB), it is essential to investigate resistance mechanisms, which can either be primary (for non-responders) or secondary (for those who develop therapy resistance after initial response). Samir Khleif investigated and defined mechanisms of resistance, including immunotherapy biologic incompatibility, immune-combination compatibility, and adaptive resistance. Some of these resistance mechanisms are introduced by the way the patients are being treated (iatrogenic). Many combination strategies are being investigated that include immunotherapies, particularly anti-PD-1 axis therapies. However, an important factor to consider is that the tumor microenvironment (TME) is a complex biologic system of tumor–immune interactions, and when you change the TME through one immunomodulator (e.g., ICB, radiation, vaccine) this alters the TME, and so may affect subsequent therapies. In a TC-1 mouse study, it was shown that combining an HPV E7 vaccine (to prime T cells) with anti-PD-1 was effective in cold tumors, even when the monotherapies were not. However, when the first PD-1 dose was provided before the vaccine, it abrogated the antitumor effects of the combination. When the researchers assessed the TME in this model, they found that the PD-1 pretreatment resulted in a loss of CD8+ T cell infiltration. In another study that combined anti-PD-1 with OX40 agonistic antibodies, treatment effects were also abrogated, while monotherapy with either antibody was effective. The combination treatment led to overactivation of T cells, resulting in annexin V upregulation and apoptosis. This increased cell death was also observed in the PD-1/vaccine model. When there was no antigen activation before PD-1 blockade, it resulted in PD-1+CD38hi dysfunctional T cells that would undergo apoptosis following PD-1 blockade. In vitro activation studies showed that suboptimal priming followed by PD-1 blockade resulted in CD38 upregulation on T cells. These cells were “confused”, with high expression of activation and suppression markers, as well as genes related to inflammatory and inhibitory markers, and had a very low chromatin accessibility. Similarly, in the clinic, non-responders had high levels of dysfunctional CD8+ T cells in the tumor and the peripheral blood. Therefore, in cold tumors, suboptimal priming combined with anti-PD-1 may result in dysfunctional CD8+ T cells, which may result in therapy resistance. To overcome this, depletion of CD38 may be effective, as was shown in mice after adoptive therapy. Clinical trials are now being conducted to test both sequencing of a NY-ESO-1 vaccine and anti-PD-1 therapy, and depletion of dysfunctional CD38+ T cells in combination with a KRAS vaccine and PD-1 blockade. These data demonstrating the criticality of proper priming before checkpoint inhibition are representative of the importance of understanding the dynamic biology of the individual agents to effectively utilize combination strategies.
Tumor immune environment
In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer - Juan Dubrot - The Broad Institute of MIT and Harvard.
The success of checkpoint blockade therapies, especially blockade of the PD-1 axis, has instigated hundreds of combination trials. In an attempt to provide a more systematic approach to target identification, Juan Dubrot conducted in vivo sub-genomic and genome-wide screens across 8 tumor models (5 histologies) in mice to identify new targets. Building on a previously published system utilizing Cas9-expressing tumor cells transfected with a library of guide RNAs to knockout selected genes, Dubrot injected these tumors into immune-deficient (NSG) mice and wild-type mice that were either untreated or treated with immune checkpoint blockade (ICB). Guide RNAs that were enriched in recovered tumors from ICB-treated wild-type mice relative to NSG mice identified genes whose loss promoted resistance to therapy (CRISPR-induced loss of function allowed these tumor cells to survive), such as the tumor suppressor PTEN or the programmed cell death gene CASP8. Guide RNAs that were depleted in recovered tumors identified genes whose loss enhanced killing by checkpoint blockade, indicative of potential combinatorial targets. These revealed both known, as well as novel potential targets, most of which were connected to a single model. Interestingly and unexpectedly, across models, depleted guide RNAs were enriched in genes in the interferon signaling pathway, which was further validated by single-guide knockout studies. The interferon pathway, through JAK/STAT, modulates hundreds of genes, and transcriptional analysis of the tumor cell lines with or without interferon stimulation revealed upregulation of multiple components of the MHC-I antigen processing machinery. Mechanistically, upregulation of classical MHC class I (A, B and C) could be inhibitory to NK cell function. Dubrot focused attention on upregulation of H2-T23, the gene for Qa-1, the mouse homolog of the non-classical HLA-E in humans. This lowly polymorphic MHC gene with a limited peptide repertoire and a restricted pattern of expression has a major immunoregulatory role via binding to the inhibitory NKG2A/CD94 ligands on NK and T cells. Cell depletion experiments showed that in these models, CD8+ T cells were predominantly impactful, and in vitro and in vivo studies directly demonstrated the immune inhibitory effect of QA-1 expression. Thus, systematic screening identified interferon stimulation as a pan-cancer mechanism of immune resistance in mice, acting through upregulation of the immune inhibitory non-classical Qa-1 (HLA-E in human) MHC molecule.
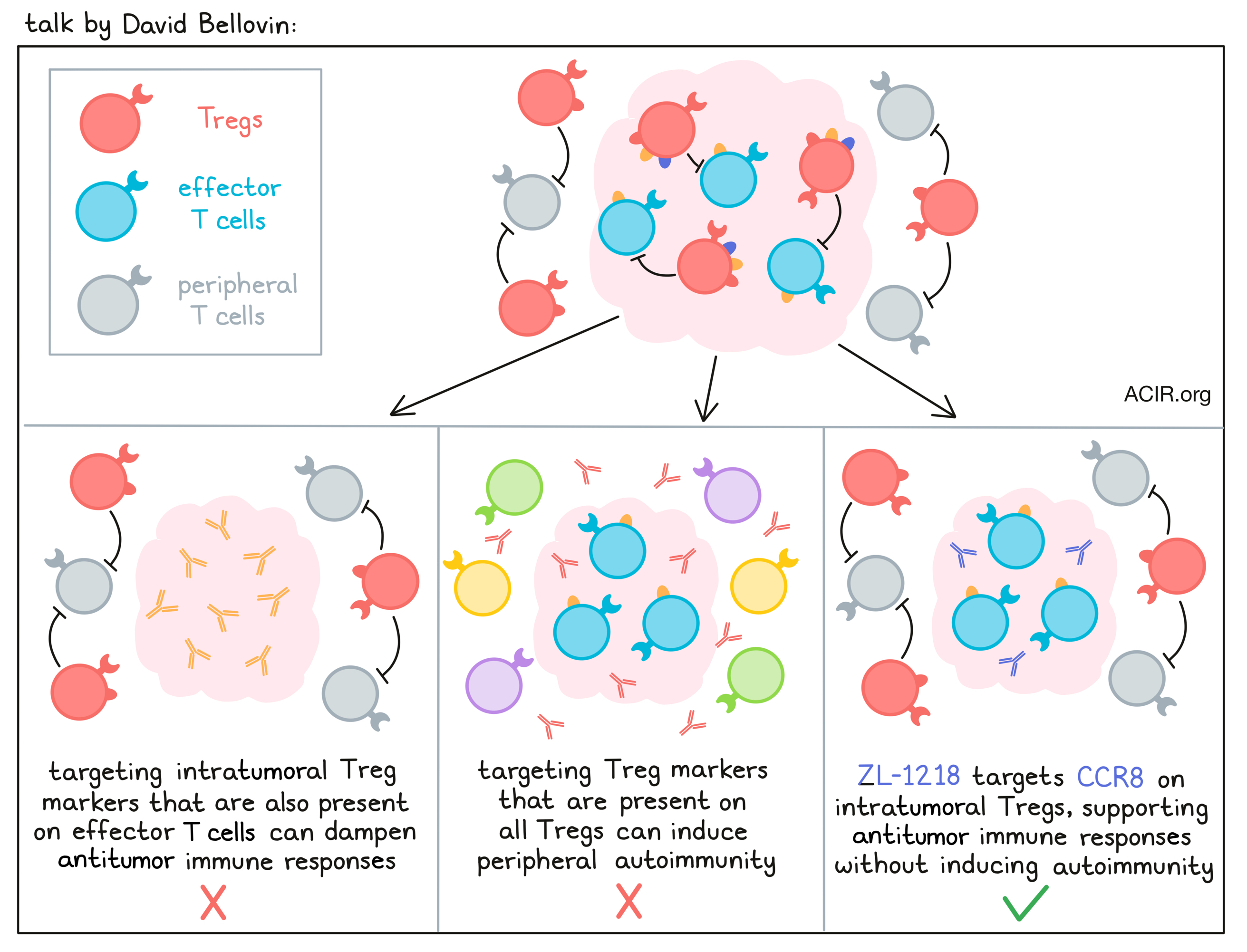
ZL-1218, a novel anti-CCR8 antibody, exerts potent antitumor effect by depleting intratumoral regulatory T cells - David Bellovin - Zai Lab.
Tregs are well known antagonists to antitumor immune responses, but targeting them in the tumor microenvironment (TME) has been a challenge, as most Treg markers are either expressed on effector cells or on Tregs throughout the body. In an effort to deplete Tregs without dampening desirable immune responses or triggering toxic peripheral autoimmunity, David Bellovin and colleagues identified the chemokine receptor CCR8 as a targetable surface marker that was consistently expressed at the RNA level in Tregs in various tumor microenvironments, with limited expression on healthy tissues, effector T cells, or peripheral Tregs. These RNA results were then validated at the protein level in multiple patient samples across histologies. Interestingly, expression of CCR8 on Tregs in the TME resembled expression on T cell leukemia and lymphoma samples. Using an extensive screening approach, the researchers then generated both murine and human CCR8-targeting antibodies that were Fc-engineered to enhance ADCC and ADCP functionality. Testing the top candidate therapeutic, ZL-1218, and backup candidates against dissociated human tumor samples, the researchers saw that the antibodies bound to target cells induced potent ADCC and depletion of cells expressing CCR8, even at relatively low levels representative of expression on Tregs in the TME. They also effectively blocked ligand binding and signaling through CCR8. In a mouse model with knock-in expression of human CCR8, anti-CCR8 antibodies depleted Tregs in the TME and induced antitumor effects in a dose-dependent manner. Further, a combination of anti-CCR8 and anti-PD-1 induced complete regressions and long-term antitumor immunity in 100% of mice. ZL-1218 also binds to cynomolgus and rat CCR8 with an affinity close to human CCR8, allowing for translatable toxicology studies, which are ongoing; no significant toxicities have been observed to date. The pharmacokinetic profile of ZL-1218 is typical for a monoclonal antibody. This and additional research has enabled Bellovin and colleagues to project a potential human starting dose and dosing regimen.
Elucidating and targeting master regulators of tumor-infiltrating regulatory T cells - Casey Ager - The University of Texas MD Anderson Cancer Center.
Tumor-infiltrating regulatory T cells (TI-Tregs) have long been known to have important immunosuppressive functions, but therapeutic targeting poses a challenge, as treatments need to maintain peripheral homeostasis, Tregs are similar to effector T cells, and mouse-to-human translation has been challenging. Therefore, Casey Ager and team set out to find new targets specific to TI-Tregs, and different from peripheral Tregs and conventional T cells. To do this, they made use of a T cell profiling dataset with four cancer types, from which these populations could be specifically sorted from both the tumor and healthy PBMCs. Using unique bioinformatic algorithms (VIPER, ARACNe), a gene expression analysis was performed on RNAseq data to examine complex regulatory networks to infer upstream regulators of differentially expressed gene signatures of TI-Tregs. This revealed a signature with 17 master regulators, with TRPS1 being consistently underrepresented. To assess which of these were functionally required, a pooled in vivo CRISPR screen was conducted. Using a sgRNA library in Cas9+ hematopoietic stem cells, master regulator-perturbed chimeras were created, and gDNA sequences of perturbed T cells were assessed to compare guide frequencies in the MC38 tumor model. This validated TRPS1 as a master regulator of TI-Tregs. Single-gene TRPS1-targeted chimeras were then created to study tumor growth kinetics in a different model (MCA205) and this confirmed more tumor immune control when this gene was not present. The researchers also conducted a drug screen of FDA-approved or investigational compounds to assess whether any of the 1554 compounds tested could antagonize any of the master regulators. Out of 195 compounds with baseline anti-Treg activity, 7 had preferential activity against TI-Tregs. In vitro, it was shown these drugs indeed inhibited the identified master regulators, including TRPS1. In vivo validation in a late-treatment MC38 model showed effectiveness of low-dose (1/10 - 1/100 of clinical dose) gemcitabine. This treatment was shown to be immunogenic and reduced TI-Treg frequencies, but did not affect peripheral Tregs, suggesting effects were, at least in part, due to TI-Treg suppression.
Intratumoral CD40 agonist sotigalimab with pembrolizumab induces broad innate and adaptive immune activation in local and distant tumors in metastatic melanoma - Salah-Eddine Bentebibel - The University of Texas MD Anderson Cancer Center.
While anti-PD-1 immune checkpoint blockade is effective in a subset of patients with metastatic melanoma, many do not respond or develop resistance. Salah-Eddine Bentebibel presented the results of a clinical trial aiming to overcome this resistance by combining intratumoral treatment with the CD40 agonistic antibody sotigalimab, with intravenous anti-PD-1 (pembrolizumab). The aim of this study was to induce local priming in the tumor by CD40 agonism, using the tumor as its own vaccine (in situ vaccination). In the phase I trial, 14 checkpoint blockade-naive patients were treated at five dose levels, and the phase II study is currently underway using the highest dose level from the phase I (the RP2D). Patients with unresectable, but measurable Stage III or IV melanoma were enrolled; some also had distant lesions. The results reported here include 30 patients treated at the RP2D as of December 30, 2021. The treatment was well tolerated and no study discontinuations have occurred. Most of the adverse events were injection-site reactions, and 20% of patients had grade 3 immune-related adverse events. Best overall response was complete response in 17% of patients, and 33% had a partial response. The objective response rate was 50%, with a disease control rate of 67%. Efficacy was seen in patients with low and high PD-L1 or lactate dehydrogenase (LDH; predictor of survival in metastatic melanoma) expression levels. Biomarker analyses were performed on tumor core needle biopsies from local and distant lesions using NanoString gene expression analysis and TCR repertoire analysis. There was an upregulation of genes associated with antigen-presenting cells and CD40 pathways at early timepoints, and T cell activation at later timepoints. There was a robust upregulation of T cells, macrophages, CD8+ T cells, and cytotoxic gene signatures in responders. Additionally, responders had an increase in Th1 gene signatures; an increase in TGF-β1 gene expression was also noted. In distant lesions, the same gene signatures were upregulated. TCRseq revealed an increase in T cell infiltration and expansion of T cell clonality. Distant and local lesions had shared expansion of new clones, suggesting the combination strategy induced broad innate and adaptive immune activation both locally and systemically.
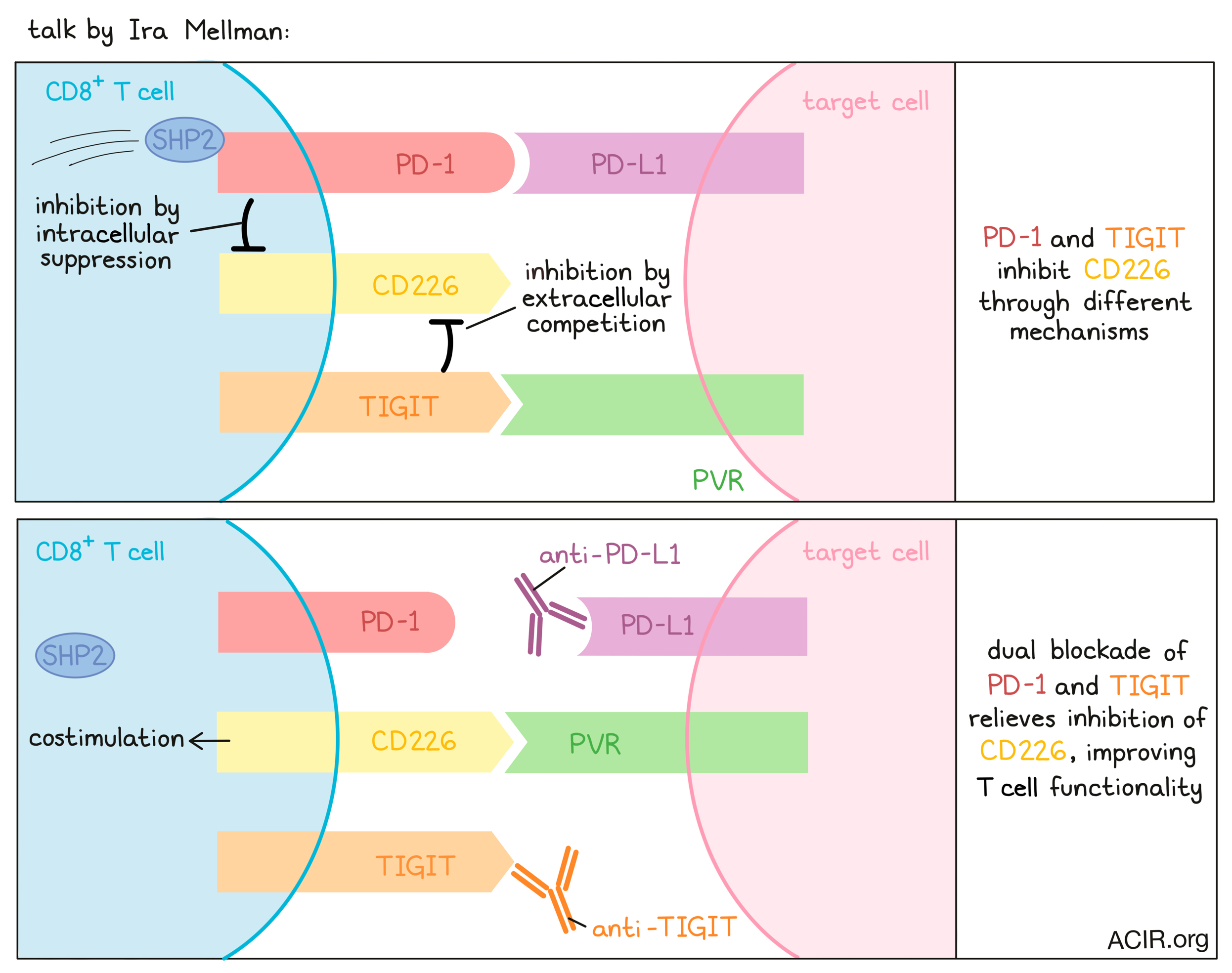
A convergence of myeloid cells and T cells controls cancer immunity - Ira Mellman - Genentech.
At a major symposium on myeloid cell control of therapeutic antitumor immunity, Ira Mellman discussed the important role these cells play, particularly in responses to checkpoint inhibitors like anti-PD-1 and anti-PD-L1. In looking at when and how these checkpoint inhibitors act on the cancer immunity cycle, it has become clear that while they may have some effect on the activity of some exhausted cells in the TME, their primary mechanism is more likely in acting early, when resting T cells are being pushed to activate, proliferate, or differentiate towards an effector phenotype. One factor pointing toward the importance of early T cell stimulation is the critical role of CD28 signaling. Tracking down the source of CD28 costimulation during checkpoint blockade, Mellman described published work linking PD-1 and CD28 signaling through the phosphatase Shp2. Evidence also showed that myeloid cells, particularly dendritic cells in the TME, were the critical source of PD-L1 ligand. Biochemical reconstitution and in vitro data have indicated that PD-1 preferentially regulates the CD28/PI3K pathway (versus TCR signaling) and that blocking the PD-1 axis may allow for stronger costimulatory signaling in T cells. Further investigation showed that PD-1/PD-L1 blockade likely acts mostly on stem-like memory T cells, which arise from naive T cells and are still fairly plastic, with the capacity to differentiate towards memory, effector, or exhausted phenotypes. In addition to PD-1/PD-L1, many researchers are also targeting a number of additional checkpoints, including TIGIT – an inhibitory receptor on T cells that is often expressed alongside PD-1. TIGIT likely works by limiting CD226 costimulation through competition for their shared ligand, PVR. Interestingly, PD-1 was also shown to inhibit CD226 intracellularly via dephosphorylation, so there is strong evidence to support PD-1 blockade and TIGIT blockade as a rational combination. There is also evidence, based on the importance of the Fc domain of anti-TIGIT antibodies, that TIGIT blockade may also induce beneficial effects in other cell types, like enhancing activation in NK cells, supporting the rewiring of Tregs into effector T cells, or activating myeloid cells through FcR clustering. An ongoing Phase II study of anti-TIGIT (tiragolumab) with anti-PD-L1 (atezolizumab) has shown promising clinical results. Interestingly, in addition to the presence of effector T cells, the presence of macrophage and Tregs have correlated with the efficacy of this combination. Finally, Mellman discussed a study that investigated what factors dictate whether a tumor will adopt an immune-inflamed, immune-excluded, or immune-desert phenotype. Using a technique that involves seeding clonal tumor cells into the ears of mice, the team was able to generate all 3 TME phenotypes, even when the mouse and tumors used were genetically identical, indicating that the tumor microenvironment plays a key role in the immune phenotype of the tumor that develops. While there is still more to understand regarding this phenomenon, Mellman strongly suspects that myeloid cells play a key role.
Myeloid programs that promote tumor immunity and tumor response to checkpoint blockade - Miriam Merad - Mount Sinai School of Medicine.
While response to immune checkpoint blockade (ICB) is mediated by T cells, priming by antigen-presenting cells is required for T cells to function, and much less is known about the role of these cells in response to ICB. Miriam Merad discussed a study in hepatocellular carcinoma in which patients were treated twice with PD-1 blockade before surgery. Then pretreatment tumor biopsies were compared to posttreatment tumor resections to map molecular responses to immunotherapy. Treated lesions showed regions that were rich, excluded, or low in T cells. Responders generally had high T cell content, while variable environments (rich to excluded) were found in non-responders. T cell-rich tumors had high levels of PD-1hiCD8+ T cells, and these cells were found in four enriched clusters: terminally exhausted, progenitor exhausted, proliferating, and effector T cell clusters. The effector clusters were dominating in responders. For CD4+ T cells, responders had an enrichment of cells with follicular helper T cell (Tfh) features, including PD-1hi, CXCL13, IL-21, IL6ST, ICOS, and CD200; these cells also expressed CH25H. The effector CD8 and Tfh-like CD4 signatures were highly correlated. Single-cell TCRseq confirmed clonal expansion of PD-1hi effector CD8+ and Tfh-like CD4+ T cells, and over 75% of these clones were found in pretreatment biopsies as well, but not in blood, suggesting local therapy-induced T cell expansion, particularly in responders. To assess what cells are involved in this process, dendritic cells (DCs) were studied. DC1 and DC2 both upregulated a regulatory program) upon capture of tumor antigens to create a novel DC phenotype, mregDC, characterized by high expression of maturation, regulatory, and migratory markers. These cells were uniquely marked by LAMP3. MregDCs were enriched in responders and were located close to Tfh-like CD4+ and progenitor CD8+ T cells. Receptor ligand analysis showed that these cells were enriched for genes associated with chemotaxis of T cells, priming and differentiation, modulation and prevention of exhaustion, and CD8 survival. These data suggest that mregDCs are essential organizers of niches that attract Tfh-like CD4+ T cells, enabling CD8+ progenitor T cell differentiation into antitumor effector CD8+ T cells. Enhancement and modulation of mregDCs may enhance responses to PD-1 blockade.
Lloyd J. Old Award in Cancer Immunology
AACR-Cancer Research Institute Lloyd J. Old Award in Cancer Immunology Lecture
As the recipient of the AACR-Cancer Research Institute Lloyd J. Old Award in Cancer, Ira Mellman discussed a number of topics relating to his broad range of research efforts over the years, from visualizing the immune synapse between T cells and their targets, to how target cells defend themselves against immune attack by using ESCRT to slough off damaged cell membrane, to the cancer immunity cycle. Discussing more current research, Mellman described how progress made developing neoantigen-targeting cancer vaccines allowed for the development of RNA-based COVID-19 vaccines, and how lessons learned from wide-scale administration of COVID-19 vaccines can feed back to fuel progress in cancer vaccines. For example, the COVID-19 vaccine formulations utilized modified single-stranded RNAs incorporating a pseudo-U, preventing the RNA from acting as an innate TLR stimulator. Instead, ionizable lipid nanoparticle packaging (compared to liposomal packaging) provided adjuvancy by inducing IL-1, which in turn controls cytokine responses. While humans and mice exhibit similar cytokine responses to this formulation, mice experience less severe side effects and can tolerate much higher doses due to an upregulation of IL-1ra, which inhibits IL-1α or IL-1β from binding to the IL-1 receptor IL-1R1. On the contrary, humans upregulate IL-1α and IL-1β, which may limit dosing.
By Lauren Hitchings, Maartje Wouters, Ed Fritsch and Ute Burkhardt.



