
Last week, the ACIR team attended the virtual AACR Annual Meeting 2021. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Tumor microenvironment and effective immunotherapy
Sergio Quezada
Theresa Kolben
Antoni Ribas
Ira Mellman
Combination therapies
Taha Merghoub
George Coukos
T cell dysfunction and exhaustion
E. John Wherry
Andrea Schietinger
Daniela Stefanie Thommen
Rachel C. Lynn
Cell therapies
Evan W. Weber (Crystal Mackall lab)
Jason Alan Chesney
Katayoun Rezvani
Christopher A. Klebanoff
Neoantigens
Frank J. Lowery III
Nina Bhardwaj
William E. Gillanders
Microbiome
Jennifer A. Wargo
Tumor microenvironment and effective immunotherapy
Targeting regulatory T cells in cancer: From mechanisms to new therapies- Sergio Quezada - University College London Cancer Institute, London, United Kingdom
Revisiting an old target with fresh eyes, Sergio Quezada discussed the potential for targeting CD25 to deplete Tregs using anti-CD25 antibodies. CD25 is the high-affinity subunit of the IL-2 receptor; it is expressed at high levels on Tregs and at much lower levels on effector T cells. When the PC61 anti-CD25 antibody failed to enhance a suboptimal combination of GVAX and anti-CTLA-4 in a mouse model, it was assumed that anti-CD25 must deplete effector cells along with Tregs, and was dropped from the pipeline. However, there was little evidence to support this theory, and when Quezada and colleagues revisited past experiments, they found that the PC61 anti-CD25 antibody had failed not because it depleted effector T cells, but because it did not deplete Tregs as intended. As it turned out, the rat PC61 (an IgG1) anti-CD25 antibody that had been used did not effectively bind to FcγRIV and was therefore unable to deplete Tregs. In an effort to restore the efficacy of these antibodies, Quezada and colleagues used Fc-engineering to convert their rat IgG1 antibody to a mouse IgG2a antibody, which allowed for binding to FcγRIV. This modified version induced a four-fold depletion of Tregs; it also induced complete tumor regression in 2/13 mice as a monotherapy, and in 11/14 mice when combined with anti-PD-1. Investigating further ways to enhance the efficacy of anti-CD25, the researchers found that it was originally designed to block the interaction between IL-2 and IL-2R, which they hypothesized might hinder IL-2 signaling in effector T cells, as effector T cells express CD25 at levels below the threshold for antibody-mediated depletion. If this were true, it would mean that the increased amounts of IL-2 present when Tregs are eliminated would be going to waste. To test this hypothesis, the team designed a version of the anti-CD25 antibody that would deplete Tregs without blocking IL-2R on effector T cells. In mice with palpable tumors, a single dose of this new antibody, anti-CD25NIB, induced complete regressions in 10/10 mice. When anti-CD25NIB was tested in an immune-cold tumor model, it induced some growth delay as a monotherapy, and provided a substantial amount of tumor control and a survival benefit when used in combination with GVAX, though mice ultimately relapsed. Mechanistic studies from this model showed that anti-CD25NIB depleted both Tregs and a population of PD-1hiCD4+ T cells (which may be immunosuppressive) and increased effector CD8+ T cell infiltration, while GVAX increased activation of effector T cells and induced some Treg infiltration. In combination, these treatments depleted Tregs, increased activated effector T cells, and increased a subset of NK cells. Based on these promising results in mice, Quezada and colleagues developed and tested the first anti-human CD25NIB. After validating its potential in vitro and in humanized mouse models, a clinical trial for anti-CD25NIB was initiated (see talk by Theresa Kolben below).
Modifying the tumor microenvironment with RG6292 (aCD25 Mab) in a Phase I clinical trial- Theresa Kolben - Roche Pharma Research and Early Development (pRED)
The anti-CD25NIB Mab described by Sergio Quezada (see talk above), and now known as RG6292, was evaluated in a Phase I dose-escalating monotherapy clinical trial and the results were presented by Theresa Kolben. The trial enrolled 21 patients with different cancer types and focused on patients with high Foxp3 expression, at least one measurable tumor, and a negative prognosis. Mild and controllable skin toxicities (primarily rash) were the predominant adverse events in the first four dose cohorts. PK analysis demonstrated a half-life of 8 to 13 days, and dose-related drug levels. Pharmacodynamics demonstrated a dose-dependent drop in Tregs in peripheral blood. Treg levels were reduced below 75% of baseline in the highest (6 mg) dosing cohort, which initiated tumor biopsies post-dosing. Histology of the post-treatment tumor revealed a strong increase of CD8+ T cells, modestly increased Foxp3+ cells, and relocation of both cell types to the inner part of the tumor, in line with an inflamed phenotype. Upregulation of PD-L1 and relocation of PD-L1+ cells to the inner part of the tumor were also observed, suggesting that RG6292 and a PD-1 axis blockade might work well in combination. A clinical trial testing RG6292 in combination with atezolizumab has been initiated.
Fighting melanoma with the immune system- Antoni Ribas - University of California Los Angeles (UCLA), Jonsson Comprehensive Cancer Center (JCCC), Los Angeles, CA
Toni Ribas presented the prestigious Presidential Address to close out his tenure as the 2020-2021 President of AACR, and focused his remarks on the mechanisms and future of cancer immunotherapy. He began with a remarkable anecdote of a patient with desmoplastic melanoma, a highly fibrobotic, collagen-rich, difficult-to-treat type of melanoma, who showed rapid tumor and fibrosis resolution upon anti-PD-1 therapy. A clinical trial testing anti-PD-1 therapy instead of surgical resection is ongoing. Ribas highlighted that these results show the power of T cells to function in even challenging tumor microenvironments. Desmoplastic melanoma was shown to have a high mutation burden, and the expressed neoantigens are known to be important to tumor control. Naturally occurring tumor-specific T cells attack the tumor and produce IFNγ. In optimal circumstances, stimulating the IFNγR on tumor cells amplifies the immune response by upregulating the antigen-presentation machinery and T cell-attracting chemokines. Unfortunately, this same pathway also upregulates PD-L1 on tumor cells, which attenuates responses. Anti-PD-1/PD-L1 checkpoint therapy can overcome this attenuation, but other resistance mechanisms exist. To uncover such mechanisms, analysis of various melanoma cell lines revealed that a subset of tumors naturally, or as a result of therapy, turn off signaling in the IFNγ signaling pathway. This knowledge stimulated ideas to overcome such blocks. One approach is to bypass IFNγ signaling by targeting pattern recognition receptors, such as TLR8 or TLR9, which sense invading nucleic acids (or mimics thereof) and stimulate Type I IFN production and signaling, similarly stimulating T cell-attracting chemokines. Clinical trials testing such approaches are ongoing. Alternatively, directly producing tumor-targeting T cells that recognize personal neoantigens is becoming feasible. In a small number of patients treated with checkpoint therapy, application of a pipeline for neoantigen-specific TCR identification showed that although the number of different neoantigen mutations that led to T cell responses in patients did not correlate with mutation burden or clinical response, the number of different TCRs recognizing these neoantigens did. Insertion of these TCRs into fresh T cells and cytotoxicity experiments confirmed the intrinsic capacity of individual TCRs from either responding or non-responding patients to kill tumor cells, supporting the importance of the broader T cell diversity observed in responding patients. Techniques using patient bulk T cells and CRISPR/Cas9 gene editing have been developed and enable multiple personal neoantigen-specific TCR approaches. Functional engineered T cells with multiple TCRs can be rapidly and effectively produced, and the T cells can be further genetically empowered by adding in engineered cytokines/cytokine receptors that can drive T cell proliferation and differentiation to desirable states (TSCM). Ribas closed by highlighting how an understanding of the basics of T cell recognition, TCR biology, and T cell signaling pathways provides the ability to induce effective immunity when natural immunity has failed.
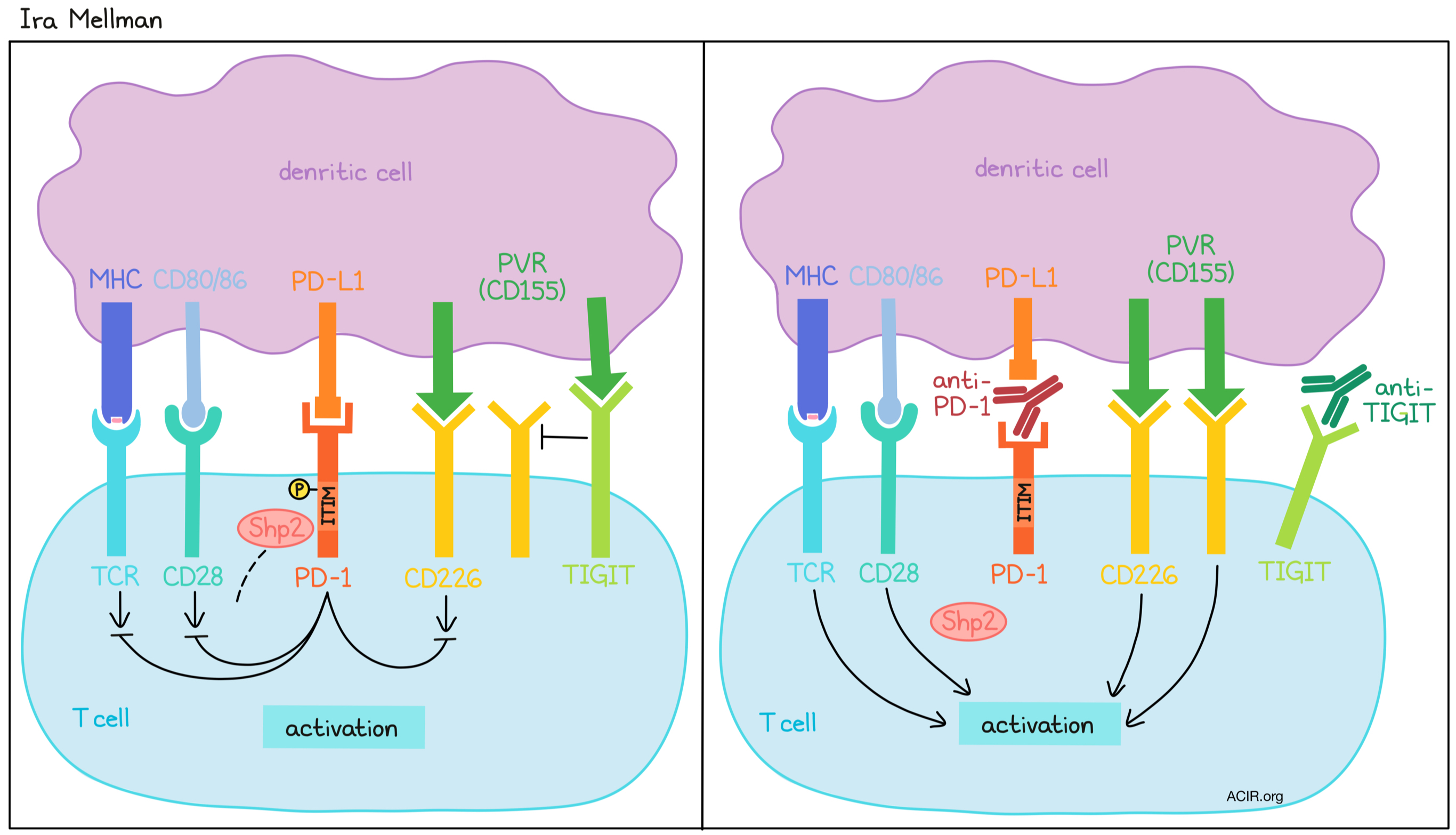
Mechanistic basis of cancer immunotherapy: checkpoints in combination- Ira Mellman - Genentech, Inc., South San Francisco, CA
Ira Mellman began by discussing the complex interactions between the surfaces of attacking T cells and their tumor cell targets. Observation of these synapses using FIB-SEM, which allows 3D reconstruction, and other advanced imaging techniques revealed a relatively smooth T cell (“synaptic”) surface and a structurally complex tumor cell (“post-synaptic”) surface with protrusions and vessels. Further, cells that have experienced membrane damage (like perforin pores) make an effort to repair it through budding, mediated by endosomal sorting (ESCRT)complexes, to remove any damaged surfaces. When ESCRT was inhibited in tumor cells, they were more susceptible to T cell attack, suggesting that this mechanism may serve as a tumor cell defense system that could potentially serve as a therapeutic target. Turning towards a more common therapeutic target, PD-1, Mellman discussed evidence supporting the idea that PD-1/PD-L1 blockade has an effect not only by limiting or reversing T cell exhaustion in the tumor, but also by enhancing T cell priming and activation by PD-L1-expressing dendritic cells in the lymph nodes. PD-L1 binding to PD-1 results in phosphorylation of the PD-1 inhibitory ITIM domain, resulting in recruitment of Shp2 phosphatase, which suppresses TCR signaling and, potentially more importantly, CD28 costimulatory signaling. Blocking PD-1 signaling during T cell activation prevents PD-1 from recruiting Shp2 phosphatase, thereby preventing this suppression and leading to preferential expansion of stem-like memory T cells that mediate an enhanced antitumor immune response, with surprisingly little effect on the exhausted T cell population. Given that these stem-like memory T cells coexpress TIGIT (and not other negative costimulators like Lag3 or Tim3), Mellman and colleagues investigated dual blockade of TIGIT and PD-1/PD-L1. They have found that TIGIT acts as a negative regulator of T cell activation by competing with the costimulatory molecule CD226 for interaction with a shared ligand, PVR (CD155). Interestingly, PD-1/PD-L1 blockade also enhanced CD226 phosphorylation, suggesting that CD226 costimulation is regulated by both PD-1 and TIGIT, a convergence that provides even more rationale for targeting them in combination. Further, anti-TIGIT is the first checkpoint inhibitor to yield positive Phase 2 clinical trial results in combination with anti-PD-L1, providing a potential clinical benefit to patients with NSCLC. A Phase 3 trial of this combination is ongoing.
Combination therapies
Combining pulsatile RAF/MEK inhibition with immunomodulation to promote effective antitumor responses- Taha Merghoub - Memorial Sloan Kettering Cancer Center, New York, NY
Taha Merghoub discussed how most therapies for cancer that are currently under investigation fall into the categories of targeted therapy or immunotherapy. Targeted therapies, like those that kill cancer cells by targeting pathways related to driver mutations, often work in a large portion of patients, but ultimately relapses occur. Immunotherapies, on the other hand, tend to induce responses in a smaller subset of patients, but the results are long-lasting. It is important to note, though, that targeted therapies can also impact immune responses by acting as in situ vaccines or altering immune-related pathways, and these effects could potentially be enhanced in combination with immunotherapy. Investigating this, Merghoub and colleagues wondered whether they might be able to enhance the antitumor efficacy of immunotherapy by combining it with downstream MEK inhibitors in the treatment of KRAS-mutant lung cancer. When administered continuously, MEK inhibitors increased MHC-I and MHC-II expression by tumor cells and induced tumor cell apoptosis, but also increased expression of tumor anti-PD-L1 and limited activation in T cells. To take advantage of the benefits, while limiting effects that might hinder antitumor immunity, Merghoub and colleagues hypothesized and showed in vitro that pulsatile treatment, rather than continuous treatment, with MEK inhibitors could induce apoptosis in tumor cells with less negative impact on T cell activation. When activated T cells were exposed to pulsatile MEK inhibition, they maintained their activation, proliferation, and IFNγ response to antigen-specific stimulation better than T cells exposed to continuous MEK inhibition. In KRAS-mutant lung tumor-bearing mice exposed to pulsatile MEK inhibition versus continuous MEK inhibition, tumor control was enhanced and TILs showed higher expression of PD-1 and CTLA-4, suggesting that the T cells were able to maintain their activation status. In combination with anti-CTLA-4, pulsatile MEK inhibition provided a slight survival advantage, which was found to be dependent on an adaptive immune response. In another set of studies, Merghoub and colleagues found that T cell function dampened by MEK inhibition could be rescued by providing costimulation with agonist anti-GITR or agonist anti-OX40 antibodies. In Lewis lung carcinoma-bearing mice, the addition of agonist anti-GITR conferred an additional survival benefit in combination with pulsatile MEK inhibition and anti-CTLA-4. The researchers have also recently generated workable models to study KRAS G12C-specific inhibitors in combination with immunotherapy, with results pending.
Mobilizing antitumor immunity- George Coukos - University of Lausanne (UNIL), Lausanne, Switzerland
The presence of intratumoral T cells directly in the tumor bed or within the surrounding stroma (“inflamed” or “excluded”, respectively) is a necessary, but not sufficient hallmark of response to checkpoint immunotherapy in patients and in animal models. Thus, approaches to effectively increase active TILs have great potential. In animal models, high-dose fractionated radiotherapy has demonstrated encouraging abscopal effects, but similar results are elusive in humans, including in a Phase II study of anti-PD-1 in combination with radiotherapy. Reasoning that an alternative strategy of tolerable, lower-dose radiotherapy to all tumor sites within a patient might provide a broader inflammatory effect and not require abscopal responses, Coukos and colleagues first investigated low-dose, whole body irradiation alone in “cold” ID8 ovarian cancer-bearing mice, which demonstrated increases in CD8+ T cell, CD4+ T cell, and CD11b myeloid cell infiltration, an increase in the CD8+/Treg ratio, and dependence on interferon activation. Moreover, upregulation of CD40, PD-1, and CTLA-4 suggested a window of opportunity for a combinatorial treatment regimen. Combining whole body radiation with multiple immune modulators (anti-PD-1, anti-CTLA-4, agonist anti-CD40, and low-dose cyclophosphamide) produced over 50% complete responses with long-term survival, and “take one out” studies demonstrated the necessity of each component. CD8+ and CD4+ T cells were significantly expanded in tumors and Tregs were reduced. Transcriptomic analysis of effector cells revealed signs of polyfunctionality, exhaustion, memory, and cytolytic capacity in both CD4+ and CD8+ compartments. Both CD4+ and CD8+ cells were required for antitumor activity. These results inspired a Phase I clinical study in a challenging “immune desert” patient population. Of 8 TIL-negative patients treated with low-dose irradiation, anti-PD-1, anti-CTLA-4, low-dose cyclophosphamide, and aspirin, 2 patients showed stable disease and 3 patients demonstrated an objective response. Transcriptomic analysis of responding and non-responding lesions were consistent with markers and cell types reflective of immune activation and suppression, respectively, confirming the results in the mouse and providing encouragement for future studies of tolerable low-dose radiation as the basis for in situ vaccination.
Back to Top
T cell dysfunction and exhaustion
What do we mean by "T cell exhaustion," and why is it important for tumor immunity?- E. John Wherry - University of Pennsylvania, Philadelphia, PA
Stepping back to discuss T cell exhaustion on a conceptual level, John Wherry re-characterized T cell exhaustion as an adaptation to frequent or constant stressful stimulation. While T cell exhaustion can be a discouraging feature in the context of immunotherapy, it is likely something that provides an advantage in certain settings. For example, exhausted T cells have been shown to contain a subset of cells that are constantly dividing in vivo, and the expression of PD-1 may play a role in preventing excessive stimulation and burnout in order to allow for this continued proliferation and long-term cellular maintenance. Further, not all T cell types can even become exhausted. Sorted, certain effector (KLRG1+CD44+) populations simply die off over a month of expansion under exhaustion-promoting conditions. Among non-effectors (KLRG1-CD44+), many cells can acquire an exhausted phenotype and persist, but certain memory populations do not become exhausted as easily as naive cells do, consistent with the increased adaptability of developmentally younger cells. T cell exhaustion is also an epigenetically distinct cell state, with highly unique chromatin accessibility patterning that is controlled at least in part by Tox. Tox promotes exhaustion while simultaneously preventing the formation of effector cells, suggesting that there is some biological benefit to inducing exhausted rather than effector T cells in some cases. Wherry presented the idea that T cell exhaustion can be thought of as a core program that can be adapted with accessory modules depending on the environmental circumstances. He also highlighted ways in which the development of exhaustion in T cells relates to other biological programs, which could help us to better understand how and why exhaustion occurs, and ultimately the purpose it serves. After positing the question of whether exhaustion is good or bad, Wherry suggested that it would be more useful to think about capitalizing on the useful, adaptive features of exhaustion while mitigating the features of T cell exhaustion that are detrimental. For example, while terminal exhaustion can limit, or even eliminate the functionality of T cells within a suppressive tumor microenvironment, progenitor exhausted T cells have useful qualities in regards to persistence and durability under the pressure of chronic antigen stimulation. Going forward, it would also be useful to better define the semantics surrounding T cell exhaustion and dysfunction, and to create a molecular atlas of human T cell differentiation.
Decoding and reprogramming T-cell dysfunction in tumors- Andrea Schietinger - Memorial Sloan Kettering Cancer Center, New York, NY
Andrea Schietinger posited the question of what molecular mechanisms drive T cells to dysfunction over the course of tumorigenesis. Investigating this, Schietinger’s team used a genetically engineered mouse model (GEMM) of liver cancer with the SV40 large T antigen as a strong oncogenic driver, which also contains immunodominant epitopes. In this model, transferred tumor-specific T cells infiltrate into lesions, become activated, upregulate inhibitory receptors, and begin to lose function within two weeks. These effects became more pronounced over time, and while early-stage dysfunctional T cells could be reprogrammed and functionally rescued (“plastic”), late-stage dysfunctional T cells could not (“fixed”). Further, these plastic versus fixed dysfunctional states were epigenetically distinct. After identifying the transcription factor Tox as an important driver of T cell exhaustion, the team looked into what drives Tox expression. They co-transferred tumor-specific T cells along with non-tumor-specific T cells into their liver cancer GEMM and saw that while both sets of cells infiltrated the tumor, only the tumor-specific cells became exhausted, suggesting that exhaustion and Tox expression are driven by antigen stimulation rather than by immunosuppression in the tumor microenvironment. When antigen-specific, Tox-deficient T cells were transferred into the same mouse model, they did not show the transcriptional or epigenetic signatures for exhaustion, but interestingly, they also did not show increased effector functions, suggesting that exhaustion programming and loss of effector functions are distinct programs that occur downstream of TCR signaling. Tox-deficient also failed to persist in these models, and were enriched for activation-induced cell death (AICD) signatures. Additional data showed that chronic TCR stimulation drives NFAT to translocate to the nucleus without AP-1, driving Tox expression and ultimately leading to exhaustion programming and upregulation of inhibitory receptors, which physiologically act as a negative feedback mechanism to prevent overstimulation and AICD, and mediate T cell persistence. In regards to what drives the loss of effector function, which appears to be uncoupled from exhaustion programming, the team noted that Tox-deficient tumor-specific cells became exhausted within tumors, while bystander cells did not, suggesting that loss of effector function is also driven by TCR stimulation, though the exact mechanism for this still requires further investigation. Finally, to address the impact of heterogeneity within TILs, Schietinger’s team investigated responses in T cells with TCRs that recognized the same antigen with different affinity strengths (by varying the epitope sequence recognized by a SV40 antigen-specific, high-affinity TCR) and found that T cells with high-affinity TCRs quickly produced maximum levels of IFNγ, but became dysfunctional very quickly. Meanwhile, T cells with low-affinity TCRs retained their effector functions for longer, but were unable to control tumors and so were functionally inert against cells expressing the low-affinity epitope. Interestingly, they were functional against cells expressing the high-affinity epitope, indicating they were intrinsically functional, but their response was epitope-dependent. This suggested that there may be a “goldilocks” range for optimal TCR signal strength and that fine-tuning signal strength could be used to enhance cytotoxic effects. In an effort to reduce signal strength in a T cell with a TCR signal that was too strong, the researchers modified the CD8α/β co-receptor, eliminating its ability to enhance affinity. This lowered the TCR signal strength and led to enhanced tumor control in vivo.
T cells in the tumor microenvironment: Exhausted, dysfunctional, or functionally adapted?- Daniela Stefanie Thommen - Netherlands Cancer Institute, Amsterdam, Netherlands
Daniela Thommen discussed the issue of heterogeneity within dysfunctional tumor-infiltrating T cells, and presented data on progresssive exhaustion in human non-small cell lung cancer. Thommen and colleagues found that TILs that expressed high levels of PD-1 (PD-1T TILs) were impaired in effector cytokine secretion and were transcriptionally distinct from TILs with intermediate or low PD-1 expression. The PD-1T TIL compartment upregulated other inhibitory checkpoint markers and expressed markers associated with tissue residency, T cell activation, and proliferation. These cells also had a higher capacity for tumor recognition, and interestingly, expressed and secreted high levels of the chemokine CXCL13, which is not typically produced by CD8+ T cells. CXCL13 is crucial for the formation and maintenance of lymph follicles, which may promote local T cell priming and have been associated with response to immunotherapy. The researchers found that PD-1T TILs were highly localized within tertiary lymphoid structures, supporting a link between the two. Looking into a possible connection between PD-1T TILs and response to PD-1 blockade in patients with lung cancer, the researchers found that responding patients had much higher numbers of PD-1T T cells within pre-treatment biopsies compared to non-responding patients, and that this was also associated with better survival. While this did not prove whether these cells could be directly reactivated by PD-1 blockade, it might indicate the presence of a tumor-specific T cell pool capable of responding to treatment. Recent studies have suggested that dysfunctional cells exist along a gradient of cell states, ranging from pre-dysfunctional to late dysfunctional, which could reflect gradual adaptation to the TME over time. To better understand dysfunctional T cell subsets, Thommen and colleagues cultured patient-derived tumor fragments, maintaining the tumor microenvironment and architecture outside of the patient. On this platform, they found that PD-1 blockade could increase T cell activation and restore effector cytokine secretion, suggesting that at least a portion of intratumoral T cells could respond to PD-1 blockade. This activation was related to a broad, early immunological response that strongly correlated with clinical responses. When the researchers blocked TCR signaling prior to PD-1 blockade, they found that much of the immunological response, including IFNγ and chemokine production, was abrogated, and that T cell activation and chemokine/cytokine expression dropped below baseline levels, suggesting that there was ongoing low-level T cell activity within tumors prior to treatment with checkpoint inhibitors. This basal T cell activity was strongly correlated with the presence of a late dysfunctional T cell subset, but not with pre-dysfunctional cells, which indicates that late dysfunctional cells may have a role in maintaining low-level tumor control. Overall, Thommen believes that exhausted or dysfunctional TILs are not functionless, but rather, display an altered function and adaptation to the tumor microenvironment, and that better understanding these varied functions and states of exhaustion could help to improve a wide range of immunotherapies.
CAR T cell dysfunction: Does exhaustion matter?- Rachel C. Lynn - Lyell Immunopharma, South San Francisco, CA
Rachel Lynn described T cell exhaustion in terms of activation-induced dysfunction. Exhausted T cells in settings of chronic infection and cancer show distinct markers, metabolic activity, transcriptomic profiles, and epigenetic profiles. One setting where T cell exhaustion is particularly apparent is in CAR T cell therapy. While this type of therapy is highly effective against many hematological malignancies, it is less effective against solid tumors, where the tumor microenvironment can be highly immunosuppressive and can induce dysfunction. Understanding exactly how exhaustion occurs could help researchers to enhance T cell therapies and improve patient outcomes. Investigating potential models for exhaustion, Lynn and colleagues found that when T cells were transduced to express high-affinity GD2-28Z CARs, the constructs would spontaneously cluster at the cell surface, inducing chronic T cell activation and functional and phenotypic hallmarks of exhaustion. Antigen stimulation, transcriptional profiling, gene set enrichment analysis, and ATACseq analysis of epigenetic remodeling confirmed the exhausted state of these cells, validating this as a strong model for T cell exhaustion. One way that this model was put to use was to investigate the hypothesis that forced overexpression of the transcription factor c-Jun could overcome exhaustion by dysregulating AP-1 activity. This strategy was found to effectively rescue IL-2 production, improve T cell functions, reduce markers of activation and exhaustion, and increase expression of genes associated with functional memory. Further, c-Jun-overexpressing GD2-28Z CAR T cells were more functional than control GD2-28Z CAR T in vivo, and mediated elimination of Nalm6-GD2 tumors in mice, conferring a survival benefit. Overexpression of c-Jun in Her2 CAR T cells also reduced exhaustion, rescued hypofunction within a suppressive solid tumor microenvironment, and improved antitumor efficacy. The GD2-28Z CAR T cell model was also used in research described by Evan Weber (see talk below), in which transient periods of rest were used to reverse or prevent T cell exhaustion.
Back to Top
Cell therapies
Transient "rest" reinvigorates exhausted CAR T cells via epigenetic remodeling- Evan W. Weber (Crystal Mackall lab) - Stanford University School of Medicine, Stanford, CA
While CAR T cells can be highly effective against hematological malignancies, their efficacy can be limited at least in part by exhaustion. To test the hypothesis that transient periods of rest could potentially limit or reverse T cell exhaustion, Weber and colleagues used GD2-28Z CAR T cells, described in more detail by Rachel Lynn (see talk above), to model T cell exhaustion. When GD2-28Z CARs are expressed in T cells, they form clusters on the cell surface, inducing chronic, antigen-independent activation and exhaustion. By incorporating a destabilizing domain into this CAR construct, Weber and colleagues created a model in which the CARs would only be expressed in the presence of a stabilizing drug, allowing expression of the CAR to be turned on and off. While “always off” CAR T cells remained healthy and functional and expressed markers of stem cell memory, “always on” CAR T cells constitutively expressed GD2-28Z CARs and became functionally exhausted (marked by expression of PD-1, Tim3, and Lag3) in as little as 10 days. Interestingly, when cells that were “on” for periods of 6 or 11 days were turned off and were allowed to “rest”, the researchers observed downregulation of exhaustion markers and an increase in stem cell memory-like markers, suggesting that rest induces functional reinvigoration of exhausted CAR T cells. Further, while “always on” CAR T cells could not control tumor growth in a cytotoxicity assay, rested cells controlled tumor growth as well as “always off” CAR T cells. Rest also rescued secretion of IL-2 and IFNγ. For comparison, the addition of anti-PD-1 to “always on” cells showed no effect. Performing RNAseq on day-15 cells revealed that “always on” CAR T cells and “always on” CAR T cells plus anti-PD-1 were closely associated, but were distant and distinct from rested CAR T cells and “always off” CAR T cells, which clustered together, suggesting that rest, but not anti-PD-1, was sufficient to limit or reverse exhaustion on the transcriptional level. To test whether epigenetic reprogramming was required for, or simply associated with reinvigoration of T cells, Weber and colleagues used drugs to block epigenetic remodeling, and found that while this had little impact on exhausted cells, it reduced IL-2 secretion in rested cells in a dose-dependent manner. Use of an inhibitor of a specific histone modification enzyme (EZH2) further suggested that epigenetic changes occurred during periods of rest that turned off genes involved in driving exhaustion. In search of pharmacological interventions that could induce rest in T cells, Weber and colleagues found that dasatinib chemotherapy, which has previously been shown to reversibly repress CAR T cell activation, was sufficient to reverse exhaustion in GD2-28Z CAR T cells, even at very late time points, with longer periods of rest inducing better exhaustion reversal. To induce rest in vivo, Weber’s team pulsted CAR T cell-treated mice with dasatinib, which enhanced tumor control and conferred a survival advantage. Ex vivo, dasatinib-treated CAR T cells expressed higher levels CD62L, had a more memory-like phenotype, and were more functional in response to stimulation.
Lifileucel (LN-144), a cryopreserved autologous tumor infiltrating lymphocyte (TIL) therapy in patients with advanced (unresectable or metastatic) melanoma: durable duration of response at 28 month follow up- Jason Alan Chesney - James Graham Brown Cancer Center, University of Louisville, Louisville, KY
Encouraged by the demonstrated antitumor efficacy of adoptive cell therapy utilizing tumor-infiltrating lymphocytes (TILs) in heavily pretreated patients with high tumor burden, Chesney and colleagues initiated a Phase 2 clinical trial investigating the cryopreserved autologous TIL product lifileucel (LN-144) in patients with unresectable or metastatic melanoma. Chesney focused on the results observed with treatment cohort 2 (cryopreserved product). All 66 patients enrolled had a high tumor burden at baseline and had previously received a median of 3 therapies, including checkpoint blockade (99% had received anti-PD-1/PD-L1, 80% also had also received anti-CTLA-4) and a BRAF/MEK inhibitor (if BRAF V600 mutant). Lifileucel was centrally manufactured in sufficient amounts, regardless of the tumor resection site, from a ~1.5 cm in diameter tumor specimen within 22 days, and cryopreserved. Patients underwent nonmyeloablative lymphodepletion (NMA-LD) prior to a one-time infusion with Lifileucel. Treatment was then followed by up to 6 doses of high-dose IL-2 over the next 2-4 days. As of December 2020, all patients experienced at least one adverse event, including thrombocytopenia, chills, anemia, pyrexia, neutropenia, hypophosphatemia, leukopenia, fatigue, hypotension, lymphopenia, and tachycardia, mostly related to NMA-LD or IL-2. Two deaths occured on the trial (one not related and one possibly related to TIL therapy). The adverse events were transient, manageable, happened early, and were consistent with the underlying advanced disease and the safety profile of NMA-LD and IL-2 regimens. At a median study follow-up of 28 months, the ORR was 36% (including 4.5% CR and 31.8% PR) and the median duration of response was not yet reached. Responses deepened over time in several patients since the prior data cut in April 2020, and one PR converted to CR. 44% of patients had stable disease. Tumor burden reductions were not associated with total cell or CD4+ and CD8+ doses. Lifileucel warrants further study in patients with advanced melanoma, and studies are ongoing in other diseases.
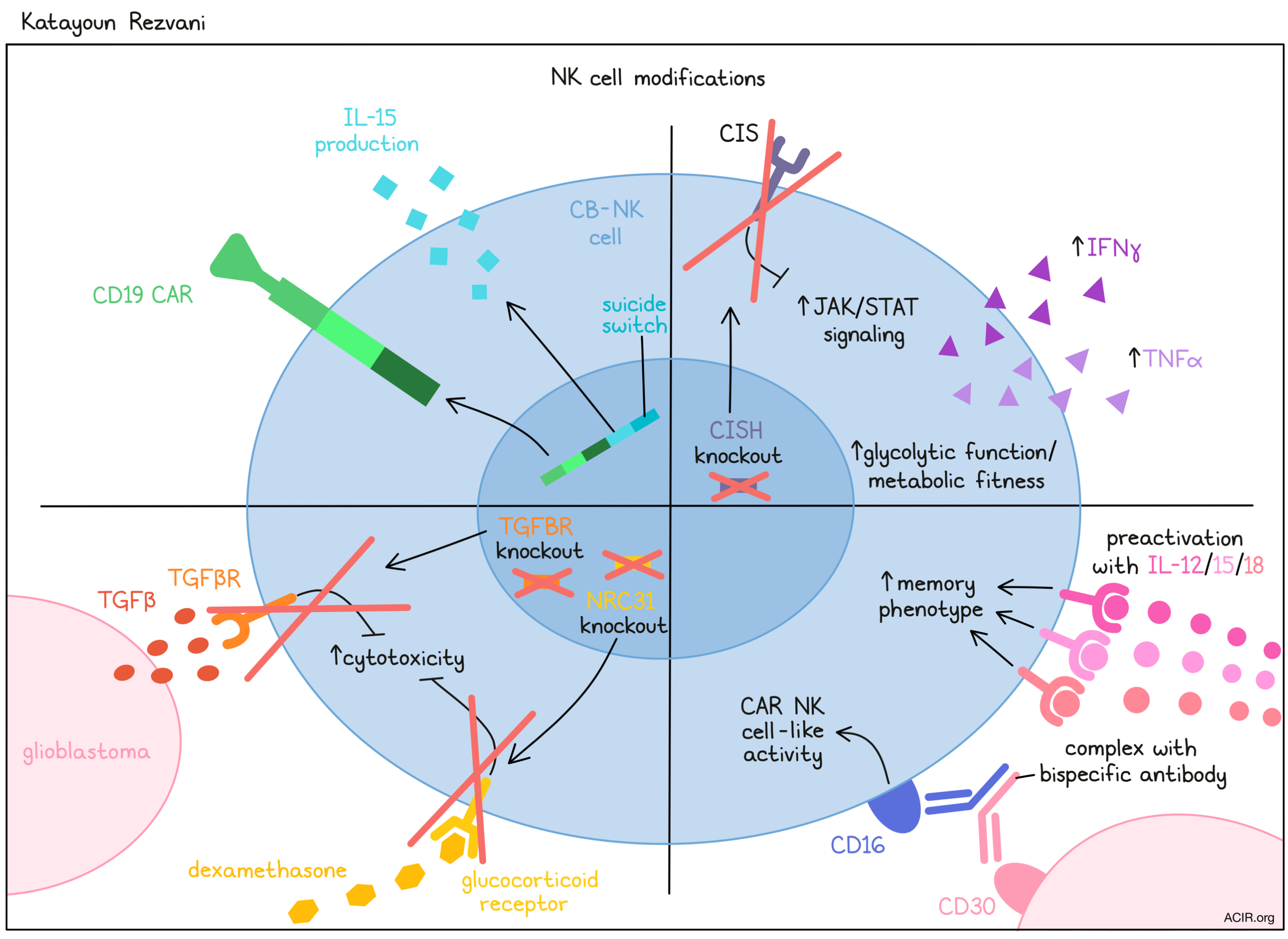
CAR NK cells: A drive to the future of cell therapy- Katayoun Rezvani - UT MD Anderson Cancer Center, Houston, TX
Building on the success of CAR T cell therapies, Katayoun Rezvani discussed the potential of using CAR NK cells as an off-the-shelf alternative. NK cells are attractive for use in cell therapy due to the fact that they are poised to recognize cancer cells, do not require antigen priming, and present little to no risk of inducing graft-versus-host disease (GvHD). There are currently nearly 20 active trials of CAR NK cells utilizing different NK cell sources and targeting different antigens. Rezvani and her team developed one CAR NK cell strategy in which NK cells derived from cord blood (CB-NK cells) are transduced with a CAR construct that is specific for CD19, induces IL-15 cytokine production, and contains a suicide switch that can be turned on in the event of unmmanagable toxicity. After showing strong antitumor effects and persistence in NSG mice engrafted with Raji tumors, a Phase I clinical trial was initiated testing the same therapy in patients. Data from this study, published last year, showed that in 11 patients who were treated, none developed cytokine release syndrome, neurotoxicity, or GVHD, and 8 experienced responses, with 7 of those achieving complete remission. NK cells expanded within patients and were detectable for up to 12 months post infusion. In another study, Rezvani and colleagues utilized CRISPR/Cas9 gene editing technology to enhance CAR NK cell functionality by knocking out CIS (encoded by CISH), an NK cell inhibitory receptor that limits IL-15 signaling. When transferred into tumor-bearing mice, these modified CISH-knockout CAR NK cells upregulated genes related to JAK/STAT signaling, increased production of IFNγ and TNFα, and prolonged mouse survival, inducing long-term cures in a proportion of animals. Interestingly, these cells also showed evidence of increased glycolytic function and enhanced metabolic fitness. Reaching for their next big target, Rezvani and colleagues extended their gene editing approach to glioblastoma, which presents additional challenges, including a solid tumor microenvironment, lack of a specific, uniformly expressed target, limited trafficking to the tumor site, standard-of-care treatment-induced immune suppression, and limited immune cell persistence within tumors. While glioblastoma stem cells (GMB GSC) can be recognized and killed by NK cells, NK cells found within glioblastoma tumors show downregulation of activating receptors, reduced cytotoxicity, upregulation of inhibitory markers, constitutive TGFβ signaling, and reduced capacity to kill target cells. Interestingly, this dysfunction was found to be induced by GBM GSC production of TGFβ upon contact with NK cells, and could be prevented by blocking TGFβ. To exploit this mechanism to enhance NK cell therapy, Rezvani’s team used CRISPR/Cas9 to knock out TGFBR2, which preserved NK cell activity in vitro and resulted in superior antitumor efficacy in vivo. Further, the team additionally knocked out NRC31, which encodes glucocorticoid receptors, thereby rendering NK cells insensitive to the immunosuppressive corticosteroids that are often prescribed to patients with glioblastoma. This work has recently been scaled up to CB-NK cells for use in the clinic, and a Phase I trial will soon begin. Finally, Rezvani discussed the possibility of getting a CAR NK cell-like effect without any genetic engineering at all. By pre-activating NK cells with IL-12/15/18 to induce a memory phenotype, and then complexing them with a bispecific antibody targeting both CD16 on NK cells and CD30 on tumor cells, Rezvani and colleagues were able to observe CAR NK cell-like activity and potent killing of CD30+ target cells. Following successful preclinical testing of this strategy in mice, a clinical trial was initiated. So far, 3 patients with refractory/relapsed CD30+ Hodgkin's lymphoma were treated at the lowest dose. No CRS, neurotoxicity, or GvHD has been observed, and all 3 patients have experienced major responses, including resolution of FDG avid disease. In addition to its clinical promise, this therapy also offers potential as a cellular therapy that is inexpensive and could be made widely available.
Targeting solid malignancies with "public" neoantigen-specific T cell receptors- Christopher A. Klebanoff - Memorial Sloan Kettering Cancer Center, New York, NY
Public neoantigens are an “elite” class of shared cancer neoantigens that are clonally conserved and derived from hotspot mutations in common driver oncogenes. Klebanoff and colleagues are systematically characterizing and discovering public neoantigens, and from this effort they elucidated the neoantigen landscape of mutant PIK3CA, the most commonly mutated human driver oncogene. They conducted mass-spec screening of the top three most recurrent hotspot mutations in PIK3CA in the context of prevalent HLA alleles to identify epitopes that are naturally processed and presented. One of these identified neoepitopes was studied in more detail. The PIK3CA (H1047L) neoepitope (ALHGGWTTK, with the hotspot H to L mutation in second position in the mutated peptide) showed enhanced thermal stability and kinetic stability in the context of HLA-A03:01 compared with wild-type. Klebanoff and colleagues then used the Memorial Sloan Kettering IMPACT clinical next-generation sequencing platform to establish an automated identification of cancer patients who express PIK3CA (H1047L) and HLA-A03:01 with automated weekly email alerts for MUT+/HLA+ patients, to isolate and characterize their T cells. The researchers detected PIK3CA (H1047L)-specific T cells from the circulation of about a third of these patients, distributed among various cancer types. The vast majority of patients exhibited clonality of the PIK3CA (H1047L) hotspot mutation and clonality was further enriched in metastatic compared to primary tumor lesions. The PIK3CA (H1047L) hotspot mutation was, moreover, clonally conserved across tumor sites within individual patients. When studying potential resistance mechanisms, no mutations in the antigen-presentation machinery were found, but HLA loss of heterogeneity was observed in 2 of 31 patients. Klebanoff and colleagues retrieved a library of TCRs that could recognize the PIK3CA (H1047L) neoepitope and cloned 5 unique TCR sequences. One of these 5 TCR sequences was identified as a lead candidate for clinical development and was characterized by superior cytolytic function with sustained cytolytic activity over time, co-receptor independence (indicative of high affinity), lack of measurable cross-reactivity, and an unusually long CDR3b region that allowed extensive interactions (H-bonds) of the TCR with the C-terminus of the neo-peptide. The developed platform enables efficient identification of public neoantigens, and development of public neoantigen-specific TCR immunotherapies for patients.
Back to Top
Neoantigens
Single cell mapping of tumor infiltrating lymphocytes enables neoantigen-reactive T cell identification in metastatic human cancer- Frank J. Lowery III - National Cancer Institute, Bethesda, MD
Frank Lowery, building on the pioneering and effective TIL therapy approach in melanoma developed by Steven Rosenberg, used advanced single-cell transcriptome and TCR sequencing to probe for neoantigen reactivity in TIL samples. Using samples from three tumor types (melanoma, colorectal, and breast cancer), Lowery and colleagues began by clustering more than 50,000 T cells from 10 different patients based on transcriptomes, identifying 12 clusters. Some of these clusters were identifiable based on canonical markers, while other subsets were less clear and were identified based on the most highly and differentially expressed gene. Clonal TCR expansion was most predominant in the CD8.TRM (C4), CD8.CCL3 (C6) and CD8.EM (C7) clusters. C4 contained “public” viral specificities. Within C7 were cells that had expanded in the peripheral blood prior to their entry into the tumor, while other clusters contained cells that were either only expanded in the tumor, or in both the blood and the tumor. These dual-expanded T cell clones have recently been shown to predict good response to checkpoint blockade. As these patients had previously been shown to have neoantigen reactivities with sequenced reactive TCRs identified, this information could be used to identify CD4.IL6ST (C1) and CD8.CCL3 (C6) as containing these neoantigen-reactive cells. Randomly selecting additional paired TCRs from these clusters (renamed as NeoTCR4 and NeoTCR8) and screening led to additional clonotypes reactive to the initially identified neoantigens. Overall, across multiple samples, 88% of identifiable neoantigen reactivity resided in these two clusters. Taking a step further, Lowery developed a unique signature from C1 and C6, which was reminiscent of dysfunctional cells (PD-1+, CD39+, Lag3+, Tox+) and could be used to prospectively identify putative neoantigen-reactive cells in three additional patients. All three patients had signature-positive CD8+ cells, while two had also positive CD4+ T cells. Screening 56 of the TCRs from these signature-positive cells demonstrated reactivity of 26 of these TCRs to shared drivers (6; recognizing two different KRAS neoepitopes), private neoantigens (8), or tumor PDX lines (10). This indicated a high rate of correspondence between cells containing the gene signature and tumor reactivity, and enabled a strategy to rapidly identify candidates for patient-specific engineered-TCR T cell therapy.
Personalized cancer vaccines: Antigen selection and translation into the clinic- Nina Bhardwaj - Icahn School of Medicine at Mt Sinai, New York, NY
Targeting the many sources of neoantigens has led to multiple clinical trials and strategies to identify optimal targets. Nina Bhardwaj presented results of an ongoing trial, along with the groundwork for planned trials spanning personal and shared (“public”) neoantigen strategies. Based on an open source platform for target neoantigen identification, Bhardwaj first described the initial results of a personal neoantigen cancer vaccine trial in the adjuvant setting in patients with a variety of malignancies, including high tumor mutation burden head and neck and lung cancer, and low mutation burden breast cancer, urothelial cancer, and multiple myeloma. In the 13 vaccinated patients, 50-90% of the immunizing peptides (7-10 immunizing long peptides per patient) were immunogenic. Clinically adverse events were low, the vaccine appears to be immunogenic, and encouragingly, 8 of 11 patients who received their entire 10 vaccine doses are still alive up to 30 weeks after vaccination ended; 4 patients who progressed responded well to checkpoint blockade. Because cost and time for personal vaccine production are high, Bhardwaj turned her attention to identifying shared neoantigen targets that can be prepared as an off-the-shelf vaccine. After briefly describing her published work on potential shared neoantigen targets in multiple myeloma and myeloproliferative disorders (a trial with a shared, frameshifted calreticulin vaccine is about to open), she focused on demonstrating the feasibility of targeting frameshift mutations in microsatellite instability-high (MSI-H) cancers (endometrial, stomach and colon cancers), where frameshifts due to small indels induced by defective DNA repair mechanisms create multiple potential neoantigen targets. Using selection criteria to maximize the potential benefit of a shared vaccine across patients, her team identified a set of 46 frameshifted novel open reading frames, varying in length from 15 to almost 170 amino acids, that were shared by at least 20% of the patients, and 5 targets that were shared across all 3 cancer types. To validate the utility of this set of 46 targets, Bhardwaj presented preliminary data demonstrating correlation of the presence of these mutations in checkpoint responsive patients and detection of presented peptides by mass spectrometry. They also demonstrated in vitro immunogenicity in healthy donor PBMCs, and specific T cell detection in MSI-H patients, both of which showed primarily CD8+ responses.
Neoantigen DNA vaccines in triple-negative breast cancer- William E. Gillanders - Washington University School of Medicine, Saint Louis, MO
Taking on the challenging triple-negative breast cancer (TNBC) setting, Will Gillanders described the background and early results of a Phase I study of a personalized neoantigen DNA vaccine in high-risk patients following neoadjuvant and adjuvant chemotherapy. Initial studies showed that predicted personal neoepitopes could be used to establish short-term T cell cultures from TNBC patients, which recognized both peptide-pulsed targets as well as tumor xenograft tissue (indicating presentation of the predicted neoantigen by the tumor). The adoptive transfer of such T cell cultures into xenograft-bearing mice demonstrated tumor control. To prepare for a Phase I clinical study in TNBC, Gillanders and colleagues first developed a DNA vaccine platform. DNA, which is relatively easy to produce, is safe and flexible, and intrinsically stimulates innate immunity, offers multiple advantages as a vaccine platform. Preliminary studies in mice with viral and tumor-associated antigen epitopes demonstrated that a polyepitope DNA vector with up to 20 epitopes separated by spacers (to eliminate novel junctional epitopes) delivered to skin and muscle cells with electroporation was feasible and immunogenic. The addition of a mutated ubiquitination domain improved degradation of the polyepitope product and antigen presentation, and prophylactic tumor-identified neoantigen vaccination in conjunction with anti-PD-L1 demonstrated antitumor efficacy in mice. Turning to the Phase I study, sequencing analysis revealed a median of 49 coding mutations per patient, with about half of these being expressed. A median of 8 strong predicted binders, along with some epitopes from driver mutations or MHC class II epitopes, resulted in a median of 10 targeted epitopes per patient. 18 patients in the Phase I, high-risk TNBC adjuvant vaccine study received 3 doses of vaccine, separated by 3 weeks (based on this preliminary work), with minimal adverse events and immune responses to the vaccine in 16 of the 18 patients. Both CD4+ and CD8+ T cell responses were observed, and most responses were mutation-specific. Preliminary TCR sequencing demonstrated dramatic expansion of some clonotypes (with de-orphanizing to determine antigen specificity in process). Although not powered to evaluate clinical efficacy, overall survival in this group was better than predicted based on historical controls.
Back to Top
Microbiome
The microbiome and cancer: Minimizing the hype and illuminating the hope- Jennifer A. Wargo - UT MD Anderson Cancer Center, Houston, TX
With a healthy dose of realistic expectations, Jennifer Wargo discussed the potential for utilizing the microbiome to modulate responses to cancer therapy, including immunotherapies. While remarkable progress has been made in recent years, not all patients respond to immunotherapy, and many experience toxicity. One of many factors influencing these outcomes appears to be the microbiome of both the tumor and the gut. The contributions of microbes to antitumor activity were first discovered when “contamination” of a stromal cell line in tumor cell co-culture studies was shown to confer tumor resistance to gemcitabine chemotherapy; co-targeting these microbes in combination with chemotherapy treatment could cure mice of their previously resistant tumors. However, not all microbes associated with tumors are bad. In some studies, high microbial diversity within tumors was associated with increased immune cell infiltration and longer survival. In another recently published study, microbial neoantigens that contribute to antitumor immune responses were observed. This highlights the importance of understanding the contributions of distinct microbial signatures and targeting them accordingly. In addition to microbes within tumors, microbes in the gut have been shown to play a role in antitumor immunity and responses to immunotherapy, and can be modulated in several ways to improve outcomes. In a recent study, fecal microbial transfer (FMT) from patients with melanoma who responded to immunotherapy into patients who did not, overcame resistance in a portion of the recipients. The gut microbiome has also been modulated in a way that reduces immunotherapy-related toxicity; in patients with immunotherapy-induced colitis, FMT from healthy donors resolved symptoms of colitis and shifted the inflammatory environment in the colon from CD8+ T cell-dominated to Treg-dominated. Other strategies to alter the gut microbiome include targeting detrimental microbes through antibiotics/phage, administering microbial consortia and probiotics, and altering the gut microbiome through diet and supplementation (prebiotics). In many scenarios, taking antibiotics just prior to, during, or after cancer treatment has been shown to have a negative impact on response and survival. Meanwhile, patients who maintain a high-fiber diet have been shown to have more diverse gut microbiomes, a higher abundance of response-associated bacteria, and better responses to anti-PD-1. In one analysis, even patients with a favorable microbiome signature benefited from a high-fiber diet. As Wargo put it, “it’s not enough just to have a good signature – you still need to be feeding it the right things”. Currently there are no dietary guidelines for patients with cancer and data is limited, however, many patients explore different dietary strategies on their own, which could impact their responses to treatment. Going forward, it would be useful to conduct dietary intervention trials in combination with treatments, taking an integrated approach. Ultimately, many personal factors, including both intrinsic host factors, lifestyle factors, cancer factors, and treatments impact the success of cancer care. It is a complex system with many variables at play, but the microbiome does play a key role and should be thoroughly investigated, as there is certainly potential to learn and improve in this field.
Back to Top
By Ute Burkhardt, Ed Fritsch, and Lauren Hitchings



