
The ACIR team attended the Keystone Symposia on Cancer Immunotherapy: Clinical Lessons to New Modalities held on March 16-19, 2025 in Banff, AB, Canada. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
(This Keystone Symposia conference is available for On Demand viewing! Find out more here).
Keynote address
Carl June
Ton Schumacher
Cytokine-based approaches
Trevor Frederick
Joanna Groom
Howard Kaufman
Jamie Spangler
Neoantigen-based approaches
Cansu Cimen Bozkus
Ana Marcu
Tumor immune microenvironment
Anna Christina Obenauf
Susan Kaech
T cell response
Stefani Spranger
Keynote Address
Cell Therapies: Current State of the Art - Carl H. June, University of Pennsylvania, USA
In his Keynote address, Carl June reflected on the first patients treated with CD19 CAR T cells, many of whom had long-term remissions (10+ years). These patients still have B cell aplasia and CAR T cell persistence, which surprisingly have been CD4+ T cells. Patients with long-term B cell aplasia had elevated type 2 immunity before treatment, and the main differences detected between patients with long- and short-term B cell aplasia were in CD4+ T cells and type 2 cytokines. In murine experiments, type 2 CAR T cells had better engraftment and memory responses. These data may lead to a new paradigm, June said, in which type 2 immunity may be considered beneficial in cell therapies.
June then discussed work on CD19 CAR T cells secreting IL-18. These CAR T cells were tested in a trial with patients in whom previous CD19 CAR T cell products had failed (but tumors remained CD19+). The study showed an 80% response rate and a 70% 2-year overall survival. This efficacy was found to be due to enhanced CAR T cell function and extrinsic effects, including the recruitment of macrophages and NK cells.
Moving to another construct, June noted that while T cells produce IL-9, they do not express the IL-9 receptor, though they can signal IL-9 through the IL-2 receptor. To improve IL-9 signaling, June’s lab created anti-MSLN and anti-PSMA CAR T cells expressing the IL-9 receptor, which were effective in tumor models. Chronic exposure assays with these CAR T cells showed that CD8+ and CD4+ cells expressed more STAT4, had higher proliferation, and showed reduction of exhaustion phenotypes.
Next, June reflected on work in which CRISPR-Cas9 was used to knock out the TCR and PD-1 genes and insert a lentiviral vector for a TCR targeting NY-ESO-1. This was feasible, although translocations were detected in 5% of the final product. Jennifer Doudna’s lab could not reproduce their data and found chromosome loss in 5-10% of cells. Their protocols differed, as June’s lab first rested the cells with cytokines before electroporation, while Doudna’s lab activated the cells with CD3 and CD28 before electroporation. Comparing the protocols, a 1000-fold difference in p53 levels was detected in the cells, suggesting the protocol order is important, and that p53 is required to preserve chromosomal integrity. June said that multiplex editing will likely be required to enhance the efficacy of cell therapies, particularly for solid tumors.
To test the boundaries of T cell gene editing, June’s lab targeted 20 different genes, base editing cells with pools of 5 genes each. Editing efficiency did not drop with increased pool sizes, and editing up to 20 genes was possible. Assessing the edited human anti-MSLN CAR T cells in NSG mice showed lethality when cells were highly modified. The cells proliferated autonomously (hyperproliferation), and caused vascular leukostasis and systemic immunopathology, with extensive lung damage, which was TCR- or CAR-independent.
Other ongoing work in June’s lab includes looking at toxicities in human trials of solid tumors. Patients with severe toxicities had baseline signatures with high levels of several cytokines, significant neutrophil infiltration, increased interferon type 2 signaling, and TNF. Using a murine model, researchers at June’s lab showed that when IL-1, IL-18, TNFα, and IFNγ were blocked, the mice survived, and the CAR T cells engrafted, with some antitumor activity. June closed by noting that it is an exciting time for cellular therapies, with many trials also in non-oncology fields, particularly in autoimmunity.
Dissecting T Cell Recognition of Cancer - Ton N. Schumacher, Netherlands Cancer Institute, The Netherlands
Opening with a canonical sketch from artist M.C. Escher to highlight his strategy of transitioning between fundamental immunology and clinical applications, Ton Schumacher focused on two topics: the application of immune oncology in the neoadjuvant setting, and cracking the code of TCR:pMHC interactions. Weighing the risks of upfront immune checkpoint blockade (delay in surgery, potential toxicity-induced elimination of the opportunity for curative surgery) and the upsides of unleashing the brakes on the immune system while the antigen burden is still high, Schumacher and Christian Blank demonstrated in a small trial (OpACIN) that immune responses were stronger with neoadjuvant therapy, with strong evidence for responses in resected tumors. Follow-on phase III trials in melanoma showed a remarkable overall survival improvement in melanoma, and a 100% 3-year disease-free survival (DFS) in locally advanced MMR-deficient colorectal cancer. Measurements of circulating tumor DNA suggested that this could be a good non-invasive biomarker that could delay or eliminate surgery. Overall, the remarkable results in neoadjuvant immunotherapy prompted Schumacher to suggest that concerted efforts should be undertaken in this setting to test molecules with good safety profiles and hints of activity in late-stage diseases.
Pivoting to fundamental immunology, Schumacher then described a strategy to understand features of relevant tumor antigens, especially given the observation that these mostly arise due to private mutations in patients, and hence require highly scalable technologies – a “Grand Challenge” for the field. To solve this problem, Schumacher described PAIR-Scan, a scalable technology that combines high complexity TCR library screening (typically used to identify TCRs reactive to single epitopes) with high-complexity epitope library screening (typically used to identify epitopes reactive with a single TCR). PAIR-Scan intersects these approaches by finding and sequencing doublets of epitope-expressing, patient-specific engineered B cell APCs and TCR-engineered immortalized reporter T cells. In screens of tens of thousands of pairs, PAIR-Scan was able to efficiently identify epitope–TCR pairs, and monitoring Ca2+ activation in the T cells improved the sensitivity. Identified pairs were regularly validated in secondary assays, were reproducibly found in duplicate screens, and the extent of enrichment (presumably a measure of avidity) correlated with cytolytic activity. Interestingly, testing TCRs from the VDJdb database with their annotated epitopes led to many failures, indicating the importance of creating a “purged” database with more confidently validated members.
Finally, based solely on sequence information of known reactive and non-reactive TCR:pMHC pairs, modeling of TCR:pMHC interactions using AlphaFold3 and examining intra-complex surface interactions using AlphaBridge demonstrated good reliability in precision-recall analysis, supporting this approach as a potentially valuable new frontier for immunotherapy.
Cytokine-based approaches
Elucidating The Mechanisms Of Novel Type I-Type III IFN Fusion Proteins For Cancer Therapy - Trevor E. Frederick, Rutgers University, USA
Aiming to tap synergistic activity between type I interferons (such as IFNα and IFNβ) and type III interferon (IFNλ) Trevor Frederick began by outlining the similarities and differences in the signalling effects of the type I and type III interferon receptors (IFNAR and IFNLR). Both receptors follow the canonical pathway involving Tyk2 and Jak1, phosphorylation of STATs 1 and 2, and nuclear transport of the IRF9/pSTAT1/pSTAT2 complex as a transcription factor activating a variety of Interferon Stimulated Genes (ISGs). ISGs affect a variety of important immune activities, including DC cross-presentation and migration, T cell cytotoxicity, M1 macrophage polarization, NK activation, and MHC upregulation. However, two key differences exist. First, the strength of interaction of typeI IFNs to the IFNAR receptor is high, and so results in a rapid activation, followed by a quick activation of negative regulators, which abrogates the signal. Conversely, the lower affinity type III IFN interaction with IFNLR prompts a slower increase, with more extended activity. Second, the distribution of the two receptors is different across a range of immune and non-immune populations, showing both shared (particularly on dendritic and epithelial cells) and distinct expression patterns.
Armed with this background, Frederick and colleagues hypothesized that a IFNβ–IFNλ fusion protein (IFNβλ) might provide a different (and better) kinetic profile of stimulation (rapid increase, but longer persistence) and expand the breadth of responding cells while synergizing on cells sharing both receptors. In vitro, mouse lung epithelial cells (expressing both receptors) showed synergistic STAT1 phosphorylation upon treatment with IFNβλ compared to a mixture of the cytokines. In an in vivo tumor control model with E0771 cells engineered to secrete one or both cytokines or IFNβλ, tumor growth suppression and eradication were significantly higher when secreting IFNβλ.
Given the risk of toxicity for systemic cytokines, Frederick developed an approach to target IFNβλ to the tumor. Knowing that the “eat-me” signal, phosphatidylserine (PS), gets translocated from the inner to the outer plasma membrane in dying or stressed cells, and that a systemically delivered antibody to PS very effectively imaged tumors upon intratumoral injection, PS seemed a reasonable target. The protein Gas6 is a naturally occurring ligand for PS. Its γ-carboxylated GLA domain tightly binds PS and is linked through an EGF domain to the LG domain, which facilitates interaction with phagocytic macrophages. By replacing the LG domain with IFNβλ, Frederick hoped to direct activity of the joined cytokine to the tumor microenvironment containing tumor cells overexpressing PS. Truncated Gas6–IFNβλ bound PS in vitro, and growth of E0771 cells engineered to express the molecule was dramatically suppressed. Control experiments with knockout of the receptors indicated this was not an effect of autocrine signaling in the tumor cells, and that the response depended on host immune cells (Rag1-/- mice). As expected, RNA sequencing of Gas6–IFNβλ-treated epithelial cells demonstrated upregulation of multiple ISG-related genes, antigen processing genes, and PD-L1 (suggesting the possibility of synergy with immune checkpoint blockade). Flow cytometry confirmed upregulation of MHC and PD-L1 at the protein level, validating the therapeutic potential of Gas6-IFNβλ.
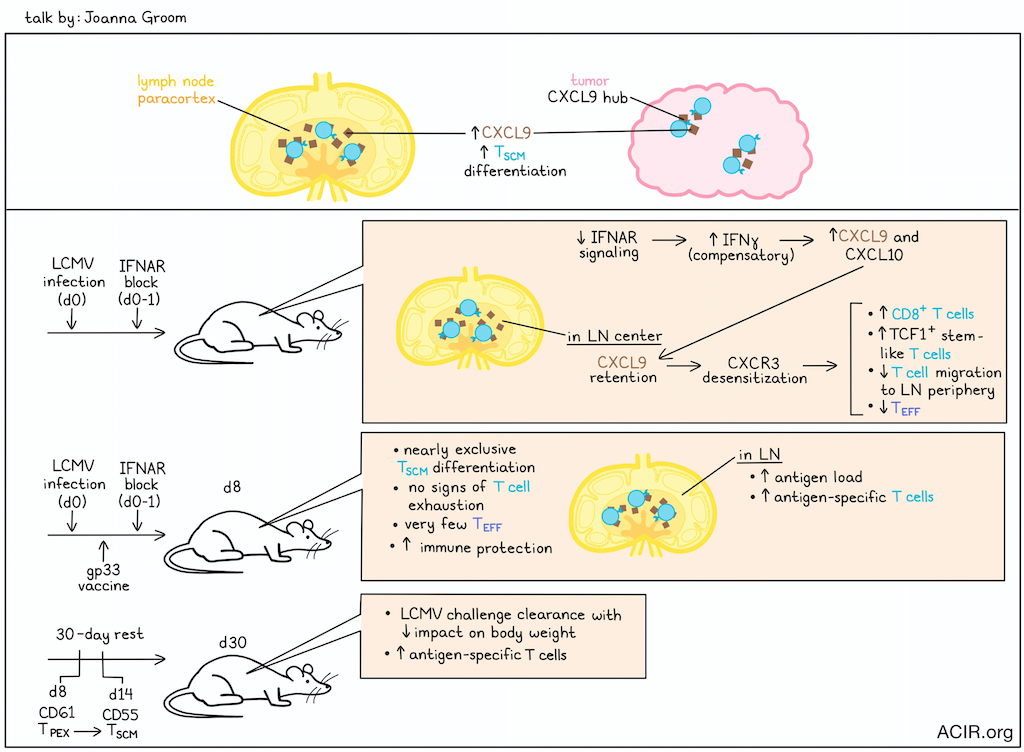
Transient inhibition of type I interferon enhances CD8+ T cell stemness and vaccine protection - Joanna R Groom, Walter and Eliza Hall Institute of Medical Research, Australia
Stem-like T cell populations have significant therapeutic potential and are crucial for vaccine efficacy. Joanna Groom explored ways to optimize the formation of stem-like memory T cells (TSCM), which provide durability in the vaccine setting, and precursors of exhaustion (TPEX), which are critical to immune checkpoint blockade. Chemokine gradients within lymph nodes (LNs) determine CD8+ T cell location and differentiation. While CXCR3 and CXCL10 drive T cell migration into subfollicular and interfollicular regions, where these T cells become effector T cells (TEFF), a CXCL9-rich niche in the lymph node paracortex, away from the inflammatory periphery of the lymph node, offers a protective environment for stem-like cell differentiation. Similar CXCL9 chemokine hubs supporting stem-like T cells are also found within the tumor microenvironment.
Groom and her team investigated whether altering the inflammatory milieu within the lymph nodes would affect CD8+ T cell differentiation. They discovered that early transient blocking of type I interferon by IFNAR block between day 0 and 1 of an acute LCMV infection (during T cell priming) resulted in a substantial increase in TCF1+ stem-like T cells and loss of TEFF differentiation in mice. Notably, IFNAR blocking did not result in chronic infection, and studies with Ifnar-/- T cells confirmed that the effect of early IFNAR block was due to environmental cues rather than T cell-intrinsic effects. Counterintuitively, blocking IFNAR signaling increased interferon-inducible chemokines CXCL9 and CXCL10. Imaging revealed CXCL9 retention, particularly in the center of an IFNAR-blocked LN, and a greater number of CD8+ T cells in the center compared to wild-type LNs. The abundance of chemokines led to CXCR3 desensitization, and hindered T cell migration to the LN periphery. RNAseq analysis showed increased signatures of type I and II interferons, inflammation, and IFNγ in the draining lymph nodes of IFNAR block-treated mice. Groom discovered that a compensatory increase in IFNγ drove the increases in CXCR3 ligands in the absence of IFNAR signaling in vivo. Mice treated with mRNA/LNP vaccines encoding gp33 and early IFNAR block (0-1) during an acute LMCV infection displayed near exclusive TSCM differentiation, no signs of exhaustion, very few TEFF, and increased antigen loads and antigen-specific T cells in the draining LN, conferring superior immune protection. After a 30-day rest, these mice were able to clear a chronic LCMV infection upon challenge, with increased antigen-specific T cells and less impact on body weight. Finally, single-cell RNAseq data revealed a transition from a TPEX population at day 8 to a TSCM population on day 14, accompanied by loss of checkpoint receptor expression. This led to the identification of new markers: CD61 for TPEX and CD55 for TSCM.
To understand the plasticity between these two populations, sorted CD61+ and CD55+ T cells were transferred to mice with recovered acute infection or day 1 chronic infection. The antigenic environment determined their expression of CD61 or CD55, regardless of the starting population. In a recovered host, CD55 expression increased, while in the chronic setting, CD61 expression increased 5 days later. This study demonstrated that timed modulation of inflammatory and migration cues direct T cell memory formation and function, suggesting a new approach to vaccine and adjuvant design.
Anchored Cytokines for the Treatment of Cancer - Howard L. Kaufman, Massachusetts General Hospital and Ankyra Therapeutics, USA
Achieving effective cancer treatment with cytokines often necessitates administering super-physiological doses, leading to systemic toxicity and limited therapeutic value. To address this challenge, Howard Kaufman and his team linked cytokines to aluminum hydroxide, an inert metal that forms local depots and resists enzymatic degradation. In this approach, pioneered by Dane Wittrup and Darrel Irvine at MIT, an alum-binding peptide with serine residues that can be specifically phosphorylated by an intracellular kinase is co-encoded at the C-terminus of a cytokine. The phosphates then interact in an electrostatic way with the hydroxyl residues on the aluminum hydroxide, forming a stable, injectable complex that serves as a slow-release cytokine depot within tumors.
I-125 SPECT imaging in mice with subcutaneous tumors confirmed that free IL-12 rapidly leaked out of the tumor, while anchored IL-12 (mANK-101) showed 40% tumor retention at day 21, with low levels in the blood. mANK-101 also demonstrated potent therapeutic activity in mice. In the CT26 colorectal model, a single dose of 5 µg led to tumor regression and a survival advantage, even in treatment of tumors >300 mm3. This effect was also seen in other tumor models, including A20 and the poorly immunogenic B16F10 model. Additionally, abscopal responses were observed, with both injected and uninjected lesions regressing.
Gene expression profiling revealed upregulation of immune response genes, including IFNγ, T cell activating markers, and chemokines like CXCL9 and CXCL10, leading to the recruitment of CD8+ T cells to the tumor site. This was consistent with an observed increased infiltration of T cells, macrophages, and NK cells in mANK-101-treated, but not control mice. mANK-101 treatment remodeled the tumor microenvironment by shifting macrophages towards an M1 phenotype, increasing the CD8+ effector T cell population (with upregulation of PD-1 and LAG-3), increasing in TH1 CD4+ cells producing IFNγ, and decreasing Tregs. Combination of mANK-101 and anti-PD-1 led to complete responses in an abscopal MC38 model, and resistance to tumor rechallenge 100 days later. In the PD-1 and cisplatin-refractory head and neck MOC1 cancer model, mANK-101 delayed growth of 10-day-old tumors by 50% and, in combination with cisplatin and anti-PD-1, resulted in significant tumor regression, improving survival rate to 80% at 100 days. Biopsies of these tumors showed profound immune aggregates that were rich in CD8+ T cells, NK cells, and occasional B cells and macrophages, which showed a higher M1/M2 ratio. Combination therapy with mANK-101 and the HDAC1/2 inhibitor entinostat led to enhanced responses and improved survival in CT26 and MOC1 models, and increased the formation of immune aggregates with TCF-1+CD8+ T cells and cCD1s (“stemness hubs”). Promising results were also observed with neoadjuvant mANK-101 in a fibrosarcoma model.
Finally, Kaufman described a phase 1 study in pet dogs with advanced melanoma using a canine version of the anchored IL-12. The study, which involved 18 dogs with mostly oral mucosal melanoma, showed no dose-limiting toxicities. Importantly, the treatment led to upregulation of immune-related genes and the formation of tertiary lymphoid structures, indicating a strong immune response. This translated to a real survival benefit, with 50% of the dogs achieving disease control, and many surviving over two years, far exceeding the typical 3-6 month prognosis. Results from an ongoing clinical study will be presented soon.
Engineering cytokines to advance cancer immunotherapy - Jamie Spangler, Johns Hopkins University, USA
Through dimerization of homo- or hetero-receptors, cytokines trigger intracellular signaling events, culminating in transcriptional and phenotypic changes in target cells. To provide greater control of these events, Jamie Spangler discussed two protein engineering approaches applied to IL-2, a key T cell-stimulating cytokine. IL-2 binds to either of two receptor complexes: the lower affinity βγ signaling complex found primarily on effector T cells (Teff), and the 100-fold higher affinity αβγ complex found on regulatory T cells (Tregs). Importantly, the βγ complex is found on low levels on resting Teff and NK cells, while the αβγ complex is constitutively expressed on Tregs, resulting in an inherently biased activity toward Tregs.
The first approach Spangler discussed was based on an anti-IL-2 antibody (S4B6), which both obstructed binding to the α receptor subunit, and also caused the same conformational shift that occurs when IL-2 binds to the βγ signaling complex in the presence of the α receptor, so flipping the bias toward Teff cells. To prevent disassociation in vivo and the release of IL-2, causing its inherent toxicity, IL-2 was fused to the N-termini of the antibodies to create immunocytokines. The anti-murine IL-2 S4B6 antibody was then replaced with the 602 antibody, which recognizes human IL-2. This enhanced the Teff/Treg bias via mutagenesis and phage display, resulting in F10, which demonstrated the desired CD8+ and CD4+ Teff-biased in vivo activity. The F10 immunocytokine was further improved by attaching a collagen-binding domain at the C-termini, a technology shown by Jeffrey Hubbell to improve intratumoral retention. In multiple tumor models, this molecule improved tumor control, with reduced toxicity.
Spangler then turned to de novo engineering of an IL-2 mimetic molecule (a “neocytokine”) in collaboration with David Baker. Focusing on the structural information of the interfaces between IL-2 and the βγ receptor, a 4-helix molecule (in the more stable up-down-up-down configuration) was designed, and then improved via in vitro selection, which dramatically improved the Teff bias (and heat stability). The resulting molecule, Neo-2, was therapeutically active, with reduced toxicity in multiple models, resulting in a significant increase in long-term survivors. Neo-2 was not immunogenic. To further improve activity, Spangler and colleagues created a conditionally active Neo-2 by splitting the 4-helix bundle into separate protein chains (1 and 3 helices) which allowed attachment of each to separate targeting domains (scFvs, nanobodies, DARPINs). Re-assembly of the Neo-2 activity when the targeting domain is bound to a target cell expressing the two separate targets would be expected to enhance tumor specificity of the cytokine action, which was observed in vitro and in vivo. Examples included dual Her2/EGFR targets, PD-L1, and Her2/PD-L1. A nanobody targeting CD8 linked to the split cytokine helices was also active, and induced enhanced T cell expansion, demonstrating cis targeting.
Neoantigen-based approaches
Personalized Neoantigen Vaccines in Cancer Immunotherapy - Cansu Cimen Bozkus, Icahn School of Medicine at Mount Sinai, USA
Cansu Cimen Bozkus presented the results of two phase I studies on the personalized genome vaccine (PGV). The PGV is a peptide-based neoantigen vaccine, developed under Nina Bhardwaj's leadership, that has, to date, been evaluated in 37 patients with various cancer types across four phase I trials. Neoantigens are predicted using the openVAX platform, which prioritizes Class I MHC binding and expression levels during ranking. Up to ten 25-mer vaccine peptides are then selected and administered in two doses of up to five vaccine peptides each into two different limbs of the patients. In the PGV001 trial, 13 patients with multiple myeloma (n=3), head and neck cancer (n=6), breast cancer(n=1), NSCLC (n=2) or urothelial cancer (n=1) received PGV in the adjuvant setting (6 doses in the priming and 4 in the boosting phases). The vaccine could be produced for all patients and was well tolerated, with only grade 1 or 2 adverse events, which were mostly injection site reactions.
An antitumor T cell response was observed in all patients to at least one of the vaccination peptides, and 44% of all vaccination peptides were positive by ex vivo ELISpot assays. The average increase in IFNγ-producing T cells was about 16-fold. T cells preferentially recognized mutant versus wild-type peptides. In vitro T cell expansion for 1 week demonstrated polyfunctional CD8+ and CD4+ T cell responses in all patients, with 38% of peptides generating CD8+ and 68% generating CD4+ T cells. TCR sequencing revealed expansion of pre-existing and induction of de novo neoantigen-specific T cell clones. Some of these clones were long-living (detected 28 weeks after the first vaccine dose) and potentially persistent. PGV001 also induced an antibody response directed against the linear immunizing peptides in all patients, with only IgM in one patient and mainly IgG in all the other patients.
Sixty months after the first vaccine dose, 6 out of 12 patients (one was lost to follow-up) were still alive. Bozkus and colleagues found a statistically significant correlation between the T cell response, in terms of magnitude and frequency of immunogenic peptides, and the alive status (excluding two patients who died of a disease-unrelated cause). Of note, in one patient who had failed prior chemotherapy plus pembrolizumab therapy before vaccination, successful treatment of cancer recurrence post-vaccination with pembrolizumab suggested a synergy between checkpoint blockade and cancer vaccination, as observed by others.
In another trial Bozkus discussed, ten patients with urothelial cancer (4 in adjuvant setting and 6 in metastatic setting) were treated with PGV and atezolizumab (starting after the first 3 vaccine doses). The vaccine could be produced for all patients and was well tolerated, with one patient developing immune-induced hepatitis related to atezolizumab treatment. Ex vivo T cell reactivity against at least one peptide was observed in all patients, with 55% of vaccine peptides being immunogenic, and an average 10-fold increase in IFNγ-producing T cells compared to pre-vaccination. T cell activity was detected across all time points examined, and patients with some of the highest T cell responses showed no evidence of disease at the time of evaluation. Both polyfunctional CD8+ and CD4+ T cell responses were induced in all patients, with slightly higher responses in the metastatic cohort. 2 of 5 patients with measurable metastatic disease had objective responses per RECIST criteria, and at 40 months, 3 of 4 patients in the adjuvant cohort remained tumor-free, and one of 6 patients in the metastatic cohort had an ongoing response.
Subcellular peptidomics elucidate HLA neoantigen presentation - Ana Marcu, Genentech, USA
The success of private neoantigen cancer vaccines, in cases where a T cell response is detectable, has prompted the need for improved strategies to identify effective neoantigens up front. The KRAS G12D mutation presents a clear opportunity, given its high recurrence across multiple cancer types, but demonstration of presentation by the common HLA-A*02:01 allele has been elusive. Why? Ana Marcu noted that current prediction approaches are relatively blind to several key aspects of the presented peptide history, including proteasomal degradation, avoidance of cytosolic peptidases, and ER processing. She postulated that analysis of the subcellular peptidome (proteasome, cytosol, ER compartment) may give insights into better predictor development.
Based on her prior work using antibodies to pull down proteasomes and associated peptides, and an approach to pull down peptides from the ER, Marcu was able to apply advanced mass spectrometry to characterize the peptides in these compartments, as well as in the cytosol and the HLA immunopeptidome. In a cell line where the KRAS G12D HLA-A*02:01 epitope has been detected by another group, Marcu identified multiple peptides from the N-terminus of KRAS in the ER compartment, including peptides with the position 12 D amino acid. However, none of these peptides had the correct C-terminus, suggesting a precursor to this epitope was not present.
Given the importance of the proteasome in generating the C-terminus of presented peptides, Marcu then tested carefully titrated doses of inhibitors of proteasomal peptidase activities and inhibitors of the N-terminal trimming ERAP enzyme. The proteasome inhibitor studies revealed that inhibition of certain subunits (for example the β5-chymotryptic-like and β2-tryoke subunits) unveiled new cleavage specificities (for example, the β1-caspase-like cleavage) as a compensatory mechanism. These types of changes result in new epitope possibilities, and Marcu noted that because the doses of proteasome inhibitors were very low, this may have clinical translation opportunities. Returning to KRAS, some of the new peptides spanning the G12D mutation could be validated as precursors to peptides presented by multiple HLA alleles (but not by the A*02:01 allele). All together, subcellular peptidomics, especially with gentle re-shaping by enzymatic inhibitors, opens up the possibilities of better validation of potential epitopes and development of better predictors.
Tumor immune microenvironment
Unlocking immunity: Decoding and reprogramming the immune-evasive tumor microenvironment - Anna Christina Obenauf, Research Institute of Molecular Pathology, Austria
While most immunotherapies have been developed and studied in the therapy-naive setting, patients might be pretreated with targeted treatments. Anna Obenauf reflected that these pretreatments may impact the tumor microenvironment (TME) and immunotherapy efficacy. Murine model experiments showed that adoptive cell therapy (ACT) was effective against melanoma in the treatment-naive setting, but not after targeted therapy, as T cells infiltration and expansion in the tumor was inhibited. Clinical datasets also confirmed this cross-resistence in patients.
Recent evidence suggests that T cells require restimulation in the tumor to maintain effector function. To assess this, the TME of immunotherapy-sensitive/targeted therapy-naive and immunotherapy-resistant/targeted therapy-resistant were analyzed. In the immune-evasive TME, several myeloid populations were lost, including monocytes, while in the permissive TME, T cells frequently interacted with myeloid cells in inflammatory hubs. To determine whether T cell restimulation needed to be local, mice were implanted with two sensitive, two resistant, or both types of tumors on opposite flanks, and T cells were injected into only one tumor. T cells expanded and moved from one tumor to the other in the sensitive setting, while there was no T cell expansion or trafficking in the resistant setting. When both tumor types were present, T cells expanded and migrated to the resistant tumor from the sensitive tumor, and induced tumor control. cDC1 knockout mice and mice depleted of all DCs showed these effects were not DC-induced.
These results led to the hypothesis that monocytes were performing this role. Two main subsets of monocytes were detected in the tumors: a non-inflammatory and an inflammatory subset, the latter of which expressed high levels of type 1 interferons and various cytokines and chemokines. Coculturing these inflammatory monocytes with naive CD8+ T cells facilitated T cell stimulation. In the model, there was evidence of TCR activation in the inflammatory hubs in the tumors. While monocytes cannot cross-present external antigens, the inflammatory monocytes harbored large amounts of neoantigens, including on MHC acquired from the tumor cells in a process known as trogocytosis, allowing effective stimulation of T cells.
In assessing how tumor cells impact the immune-evasive TME, Obenauf and team found strong upregulation of prostaglandin E2 (PGE2) metabolism and downregulation of type I IFN signaling in these tumors. To assess whether this played a role in T cell stimulation, Ptgs1/2 (enzymes responsible for producing PGE2) were knocked out of tumor cells, and in another experiment, EP2 and EP4 were knocked out of the myeloid cells (to limit response to PGE2). Both experiments made the resistant tumors permissive to ACT efficacy. To determine the role of IFN tone downregulation, tumor cell-derived type I IFN was restored, and this also restored T cell functions after ACT. Therefore, both PGE2 and IFN-I determine myeloid cell abundance and their inflammatory states.
Finally, Obenauf and her team found that high levels of PGE2 could reduce the capacity of T cells to acquire an inflammatory state. To target these mechanisms, COX1/2 inhibitors that limit PGE2 production might be of interest. In clinical trials assessing ICB, patients who concomitantly received NSAIDs (inhibiting COX1/2) had better clinical responses. In the preclinical models, COX2 inhibition partially restored the immune permissive TME and responses to ACT, and stopping treatment quickly reversed the effects.
Reprocessing TCR signaling in exhausted T cells - Susan M Kaech, The Salk Institute, USA
Susan Kaech presented research assessing how exhausted CD8+ T cells become less responsive to antigens. Given that inhibitory receptors are upregulated during differentiation towards exhaustion, it is assumed that this inhibitory signaling dampens TCR signaling. However, Kaech and team found new ways in which TCR signals are propagated in these cells. When comparing terminally exhausted cells with progenitor cells, exhausted cells were found to be less responsive to stimulation and have reduced phosphorylation of p38, c-Fos and c-Jun, while other factors showed no differences. RNA expression and flow cytometry analysis showed that expression of PKC theta and eta changed over time as cells differentiated from progenitor to exhausted phenotypes. Exhausted T cells downregulated PKC theta and upregulated PKC eta. Knockout models showed that loss of PKC theta primarily affected (reduced) the number of T cells, while loss of PKE eta did not impact T cell numbers, suggesting that PKC theta is necessary to sustain proliferation of the exhausted population. The theta knockout also had a reduction in progenitor cells, while eta knockout did not affect this population. Over time, the eta knockouts had greater functionality than controls when chronically exposed to antigen, with lower TOX expression, suggesting PKC eta is necessary for driving phenotypes associated with terminal T cell exhaustion. Stimulation of cells in culture with PMA demonstrated degradation of PKC theta, without impacting PKC eta, and when combined with repeated antigen simulation, produced exhausted T cells, which had increased TIM3, reduced TCF7 expression, and lower cytokine production. These data suggested that PKC theta is required to initially sustain T cell responses during persistent antigenic stimulation, but gets degraded when activated, while eta is not impacted.
Using mutants to create degradation-resistant PKC theta, it was established that the K413R mutation abrogated degradation of theta, and that overexpression of the K413R mutant improved CD8+ T cell numbers and antitumor T cell functions in vivo. To determine kinases downstream of PKC eta signaling, a phosphoproteomic analysis of CD8+ T cells in different states of exhaustion and in wild-type versus KO cells was performed. Several novel kinases not previously studied in T cells were found to get activated by PKC eta in exhausted T cells. The data suggested that the way in which a T cell responds to TCR signals varies based on its biochemical makeup (i.e., composition of kinases present) and the differentiation state. Inhibition of casein kinase 1 in cell cultures resulted in more progenitor-like cells with increased TCF and decreased TIM3 expression. Further investigation revealed that the previously under-explored G2 casein kinase 1 family member had the most impact. Depletion of this kinase improved T cell numbers and antitumor functions.
To determine whether these PKCs are also relevant in human cells, Kaech’s lab assessed human GD2 CAR T cells in the in vitro exhaustion assay with added PMA, which led to less functional and exhausted phenotypes. In this assay, inhibition of CK1 improved CAR T cell functions.
T cell response
Not all T cells are activated equally: is T cell exhaustion good? - Stefani Spranger, Massachusetts Institute of Technology, USA
Stephani Spranger began by presenting the idea that T cell exhaustion might be a good thing, and that dendritic cells have an important role during priming in determining that fate. Sensitivity to immune checkpoint blockade (ICB) varies across tumor histologies, prompting Spranger to hypothesize that the contexture of the TME determines the sensitivity to ICB, and to thus set up a mouse model in which a non-small cell lung cancer cell line is implanted orthotopically into the lungs or subcutaneously on the flank. After dual ICB, flank tumors responded to therapy, while lung tumors did not, even though lung tumors had higher T cell levels prior to therapy. Using a murine tumor model with a known antigen and adoptive transfer of antigen-specific T cells (ACT), it was shown that 72 hours after transfer, T cells in each tumor differed, with >1,000 differentially expressed genes. In the flank tumor-draining lymph node (TDLN), T cells represented early exhausted cells, and in the lung TDLN, there were memory T cell responses and high IFNγ, which resulted in a highly suppressive Th1 Treg population that restrains DCs and induces T cell dysfunction.
Spranger then went on to describe an additional factor impacting the determination of T cell phenotypes in the lymph nodes: the neoantigen architecture. Clinically, sub-clonal antigen expression (tumor heterogeneity) cases are ICB-resistant, and antigens are generally restricted to one part of the tumor. In clonal cases, even though the same antigen load is present, antigens are expressed on all tumor cells, and this subtype is sensitive to ICB. In a model system representing the sub-clonal setting, in which two tumor cell lines (each expressing only one type of antigen) were implanted, individual cross-presenting DCs in the TDLN were found to have one of the antigens of the tumor, suggesting the DCs took up debris from only one cell and migrated to the TDLN. In a clonal tumor model expressing two antigens per cell, the DCs were found to be positive for both antigens. Cells engineered clonally with two different antigens also allowed Spranger to demonstrate immune dominance by T cell induction in vivo, and immunopeptidomics in vitro was consistent with an active competition between two different strong neoantigens for binding the MHC. Interestingly, the DCs changed phenotypically when two neoantigens were being presented, with more costimulatory molecules and fewer inhibitory molecules.
Spranger then tried to modulate the T cell fate decision by treating mice shortly after tumor implantation with IFNβ, anti-CD40 or anti-PD-1. All three treatments pushed T cells into more differentiated states and produced T cells with different tumor-controlling capabilities. Surprisingly, the anti-PD-1 treated cells, which appeared more exhausted, outperformed the other treatments. Similar results were observed in the neoadjuvant setting. Spranger concluded by suggesting that this priming-associated T cell exhaustion might help the T cells to survive in the harsh TME conditions.
By Ute Burkhardt, Ed Fritsch, Maartje Wouters, and Lauren Hitchings.
(This Keystone Symposia conference is available for On Demand viewing! Find out more here).



