
The ACIR team attended the Keystone Symposia on Cancer Immunotherapy: Beyond Immune Checkpoint Blockade and Overcoming Resistance held on March 17-20, 2024 in Whistler, BC, Canada. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below. (This Keystone Symposia conference is available for On Demand viewing! Find out more here).
Keynote address
James P. Allison
Resistances mechanisms
Antoni Ribas
Padmanee Sharma
Judith Agudo
Combination approaches
Thamizhanban Manoharan
Ruozhen Hu
Basic research findings
Miriam Merad
Haydn Kissick
Jyh Liang Hor
Marcus Bosenberg
Jacqueline Ling Yee
Avery D. Posey
Improving antitumor immunity
Jonathan Lim
Alexander Keller
Klaus Heger
Matthew Gubin
Methods to analyze tumor immunity
Ronald N. Germain
Garry P. Nolan
Keynote Address
Immune Checkpoint Blockade in Cancer Therapy: New Insights into Therapeutic Mechanisms of anti-CTLA4 and anti-PD-1- James P. Allison, University of Texas MD Anderson Cancer Center, USA
Jim Allison, Nobel Laureate in 2018 for his foundational contributions to cancer immunology at both the basic and the therapeutic levels, presented the Keynote Address for this Keystone Symposia, with a major focus on T cell inhibitory and activating costimulatory receptors, especially CTLA-4. Allison began with an overview of the biology of CTLA-4 and its critical role in dampening T cell responses, which get explosive when CD28 is adequately stimulated, and the groundbreaking work from his lab in recognizing the value of blocking this inhibitory receptor for cancer treatment. These efforts culminated in approval of the first modern cancer immunotherapy, ipilimumab, in 2011, delivering a “long and flat” survival curve, now emblematic of successful anti-CTLA-4 therapy. Despite success, there is still much room for improvement, suggesting there may be other relevant checkpoints, and the work of Tasuku Honjo (also Nobel Laureate in 2018) brought PD-1 into the limelight. Anti-PD-(L)1 therapy operates on different T cell subsets via a different mechanism, explaining some remarkable results of the combination therapy in melanoma. After comparing and contrasting the features of these two critical inhibitory receptors, Allison went on to describe experiments unraveling their biological effects in murine models. CYTOF analysis of MC38 tumors treated with anti-CTLA-4 or anti-PD-1 showed some unique effects of anti-CTLA-4 in the depletion of Tregs and induction of Th1-like ICOS+ CD4+ T cells, and strong induction of PD-1hiLag3hiTIM3hi exhausted CD8+ T cells by anti-PD-1. Interestingly, the combination of CTLA-4- and PD-1-blocking antibodies reduced the number of exhausted T cells, but the number of those cells were now irrelevant to tumor control, as CD8+ effector T cells and CD4+ Th1-like effectors increased. Allison then turned his attention to ICOS, another member of the crucial CD28/CTLA-4 superfamily, based on studies of clinical samples from anti-CTLA-4 treated (but not anti-PD-1 treated) patients showing stimulation of a novel ICOS+ Th1-like CD4+ T cell population. Looking for synergy in targeting both receptors, he reviewed past and recent experiments demonstrating the synergy of ICOS stimulation (via a cellular vaccine expressing ICOS ligand) and anti-CTLA-4 in terms of improved tumor control, but also biologically by remodeling the myeloid cells in the TME. ICOShiTbet+ CD4+ T cells were unique to anti-CTLA-4-treated samples, which could be recapitulated in CTLA-4 knockout mice, leading to the hypothesis that preventing CTLA-4 signaling (“negative costimulation”) generated novel CD4+ T cell phenotypes (but not novel CD8+ T cell phenotypes), and that this was perhaps related to stronger CD28 signaling and prior observations of stimulation of lower-avidity T cells. Finally, Allison described a series of experiments examining the role of CTLA-4 blockade in memory cell formation. Vaccination with irradiated GM-CSF-transduced tumor cells, irradiated wild-type tumor cells, or a peptide antigen in conjunction with anti-CTLA-4 or anti-PD-1 blockade led to dramatically different memory responses ten or more weeks later (measured by rejection of tumor challenge or levels of re-induced cells). In each case, co-treatment with anti-CTLA-4 led to stronger, more effective memory than co-treatment with anti-PD-1, which was also confirmed using adoptive cell transfer. Analysis of the cell phenotypes showed that co-therapy with anti-PD-1 led to induction of TOX+ cells, while treatment with anti-CTLA-4 led to TCF7+ cells; knockouts of these genes confirmed their biological importance. Allison closed by proposing that more basic biology needs to be done to unravel this complex milieu of stimulatory and inhibitory receptors – including seeing how one treatment impacts the biology of the perturbed immune system – to build a more rational framework for conducting clinical studies.
Resistance mechanisms
Role of IFN-gamma in Response and Resistance to Cancer Immunotherapies- Antoni Ribas, University of California, Los Angeles, USA
With a focus on using clinical samples to understand and overcome mechanisms of resistance to cancer immunotherapy, Antoni Ribas began by outlining one of the key mechanisms the few T cells attacking a tumor use to amplify their response across the tumor – production of diffusible IFNγ. Longitudinal sequencing of human tumor samples from a biomarker study before and after checkpoint blockade therapy revealed upregulation of genes related to IFNγ stimulation, including genes associated with cell cycle arrest, antigen processing and presentation, IFNγ signaling, and T cell-attracting chemokines. This upregulation was stronger in clinical responders. In the course of this therapy, the T cells were hijacking the IFNγ-responsive pathway in tumor cells to make the tumor cells more visible to the immune system and more sensitive to killing. Key signaling molecules in this stimulatory pathway include both JAK1 and JAK2, and Stat1. Prior work from the Ribas lab had shown that acquired resistance to anti-PD-1 therapy could be due to homozygous de novo loss-of-function mutations in JAK1 or JAK2, clinically confirming the importance of IFNγ signaling to T cell-activating therapies. Although a mechanistic understanding of how IFNγ signaling affected most of the identified gene pathways existed, little was known regarding how it affected cell proliferation and pro-apoptotic characteristics. A screening showed that nearly all of 31 tested melanoma cell lines were inhibited in their growth by IFNγ exposure, indicating that this was a generalized effect, supporting deeper analysis. To determine what other pathways were involved in IFNγ-induced growth inhibition, the Ribas lab conducted a drug screen (testing drugs for the ability to block IFNγ-induced growth inhibition) and observed that ERK and RAF inhibitors were the most active hits after JAK inhibitors; this was confirmed in a CRISPR screen. Although counterintuitive, given the known role of oncogenic mutations in these pathways, Ribas confirmed that IFNγ led to upregulation of phosphoERK. Inhibiting ERK with titrated levels of inhibitors that only blocked the enhanced IFNγ-induced ERK phosphorylation was able to overcome the growth inhibition induced by IFNγ and maintain cell viability – an effect that was confirmed in 17 of 23 melanoma cell lines with different driver oncogenes. To understand next what gene pathways were involved in IFNγ-mediated growth inhibition, RNA sequencing was used to determine what genes were upregulated by IFNγ, but not by IFNγ + ERKi. This revealed that stress-induced pathways predominated (some of these genes had been identified in the CRISPR screen also). Knockdown of the identified genes is currently being conducted to provide more detailed information. In summary, these results gave rise to a model in which ERK stimulation is important for maintaining cancer cell viability and growth, and ERK inhibition can drive cells toward apoptotic death, but overstimulation of this pathway causes stress responses that can also lead to cell death. Ribas closed by suggesting that in some circumstances, overstimulation of the pERK pathway could be used to enhance responsiveness to immunotherapy, and showed in vitro the synergy in induction of cell death by combination of IFNγ and a BRAF inhibitor (as an indirect way to turn on ERK).
Neoadjuvant and Tissue-based Trials for Evaluating Mechanisms of Response and Resistance- Padmanee Sharma, University of Texas MD Anderson Cancer Center, USA
Pam Sharma discussed the approach of using neoadjuvant trials and deep analysis of patient tissue to better understand why some patients respond to immune checkpoint blockade (ICB) while others do not, and to identify biomarkers that can aid better patient selection for response or toxicity. With over 3000 ICB trials ongoing, the science needs to keep up with the explosion in interest. Neoadjuvant clinical trials that are initiated by hypothesis-generated mouse studies can provide tissue samples that can allow in-depth analyses that both test the hypothesis, and generate new hypotheses for further well designed studies in mice (“reverse translation”). Examples for the utility of this paradigm came from two very early neoadjuvant trials with anti-CTLA-4 +/- anti-PD-L1 in bladder cancer, which were the first to show pathological complete responses with checkpoint blockade. These studies revealed T cell infiltration, tertiary lymphoid structure (TLS) formation as a biomarker of response, and induction of ICOS+ CD4+ T cells, which are essential for a strong response (see Keynote address by Jim Allison). Turning to mechanisms of resistance to ICB in tumors with low mutational burden, Sharma showed how a neoadjuvant/tissue analysis study in prostate cancer revealed upregulation of the inhibitory receptors PD-L1 and VISTA on macrophages – a type of phenomenon only determinable by longitudinal analysis. As another investigation into an ICB-resistant tumor type, comparison of TME profiles in murine melanoma (B16F10) and pancreatic cancer (mT4) revealed that the pancreatic tumors demonstrated significantly higher frequencies of fibroblasts marked by TSG6, an enzyme important to anti-inflammatory processes and protection from tissue damage. TCGA data confirmed higher expression of TSG6 in human pancreatic cancer than melanoma, and single-cell RNA sequencing of human pancreatic tumors and normal pancreatic tissue showed a significant increase in TSG6+ fibroblasts in the tumor. Returning to mouse models, an available blocking antibody to TSG6 led to increased survival in the mT4 model, and a decrease in suppressive macrophages in the TME. Preliminary efforts indicate that one of the ligands for TSG6 may be CD44. CD28 and TCR stimulation also have epigenetics effects, and Sharma showed another iterative example in which non-responders to anti-CTLA-4 therapy demonstrated upregulation of the epigenetic modulator EZH2 in CD4+ T cells, which stabilized Foxp3 expression. Inhibition of EZH2 in the mouse showed reduction of Foxp3 and induction of IFNγ in Tregs and, importantly, a shift from suppressive to effector functionality. Translating this back to the clinic in an ongoing multi-disease trial, some exciting tumor remissions have been observed in patients resistant to prior ICB. Correlative analysis has revealed increases in the effector T cell population and modification of the myeloid compartment in these patients. Finally, to analyze factors contributing to the toxicity of ICB, samples from patients very recently experiencing myositis or myocarditis (less than 96 hours from presentation) were subjected to scRNAseq, identifying an increased frequency of T cells and myeloid cells, the latter of which were marked by IL-1β and TNFα expression. A clinical trial is planned to assess the impact of an IL-1 pathway inhibitor to counter induction of these toxicities.
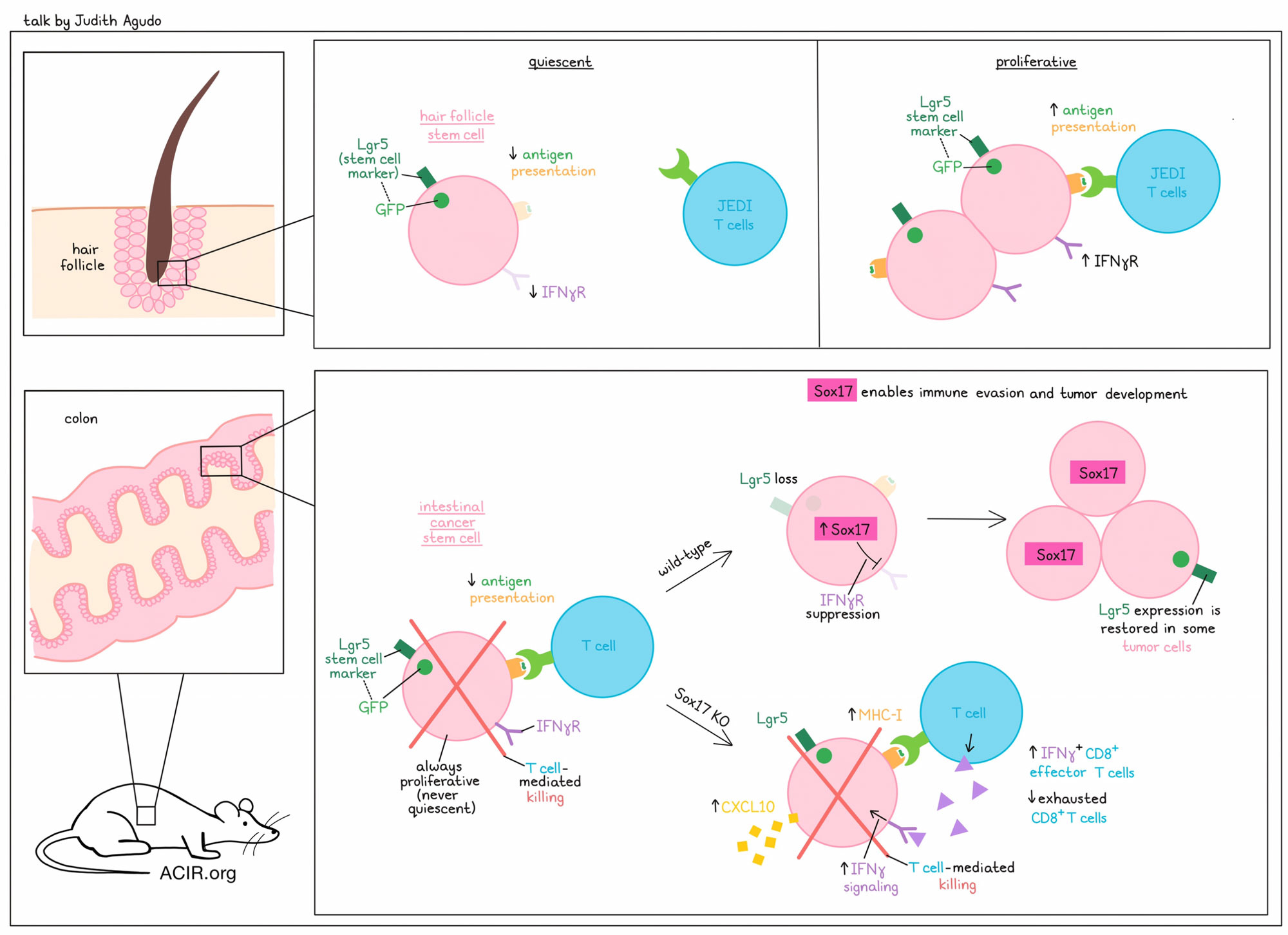
Uncovering Hidden Drivers of Immune Evasion: Stemness and Cell Cycle- Judith Agudo, Dana-Farber Cancer Institute, USA
To tackle the question of whether and how adult tissue stem cells are immune privileged, Judith Agudo developed JEDI (just EGFP death-inducing) T cells that target GFP-expressing cells. By treating Lgr5-GFP mice with JEDI T cells (Lgr5 is a ubiquitous stem cell marker), Agudo showed that quiescent Lgr5+GFP+ hair follicle stem cells (HFSC) in the skin are indeed immune privileged. Only during the restricted time frame of the anagen phase (the phase of synchronized hair growth) do HFSC start to proliferate, and this is the time when they can be easily eradicated by JEDI T cells. Genes in the antigen presentation pathway, and notably the IFNγ receptor (Ifngr1), were downregulated in quiescent HFSC, and upregulated when the cells went into cell cycle in the anagen phase. In the intestine, however, Lgr5+GFP+ stem cells are always highly proliferative and easily eradicated by JEDI T cells, leading Agudo to question how colorectal cancers could form from these highly immunogenic intestinal cancer stem cells and evade immune surveillance. Repeated colonoscopy-guided implantation of normal colon organoids harboring mutant KrasG12D with loss of p53 and APC (AKP) into the colon of recipient mice ultimately created tumor-derived AKP organoids, in which Sox17 was found by RNAseq and ATACseq to be the only upregulated gene. Sox17 is a transcription factor involved in fetal development, and while it is silenced in healthy colon tissue, it is essential for colon cancer development. Sox17 knockout showed drastic reduction in organoid tumor growth in immunocompetent mice, but not in immunodeficient mice, suggesting a potential role of Sox17 in immune evasion. Single-cell RNAseq, immunohistochemistry, and FACS analyses showed increased infiltration of effector-like IFNγ+CD8+ T cells, and almost no exhausted CD8+ T cells in Sox17 KO compared to wild-type tumors. CD8+ T cell depletion rescued growth of Sox17 KO tumors. Furthermore, Sox17 KO induced enrichment of interferon gene signatures, increased IFNγ signaling via upregulation of Ifngr1, and increased CXCL10 and MHC I in colon organoid cultures. Further experiments showed that Sox17 directly repressed the IFNγ receptor expression in these organoids, and Ifngr1 deletion rescued the tumor growth in Sox17 KO tumors. Since colon cancers originate from adenomas, the researchers crossed Lgr5-CreERT-GFP mice with APCf/f mice to investigate the role of Sox17 in adenoma formation, and showed that deletion of Sox17 eliminated the appearance of adenomas. Sox17 is highly expressed in adenomas, but is mutually exclusive with Lgr5. To rule out a vaccinal effect, knockout of Sox17 prevented the formation of adenomas in a CD8+ T cell-dependent manner. Analyses of around 30 human adenoma samples confirmed Sox17 expression in patients as well. Advanced human colon adenocarcinomas with high Sox17 expression demonstrated low levels of infiltrating CD8+ T cells, while carcinomas with low Sox17 levels contained high numbers of CD8+ T cells. In conclusion, during active immune surveillance, highly immunogenic Lrg5+ cells that acquire oncogenic mutations are eliminated by the immune system. However, during tumorigenesis, some intestinal stem cells can upregulate Sox17, leading to oncofetal reprogramming of these cells with downregulation of Ifngr1, IFNγ signaling, and MHC-I, allowing them to evade immune surveillance. Once the tumor mass and an immunosuppressive tumor microenvironment has formed, Lgr5 is turned back on in some cells, allowing them to form metastases and contribute to tumor recurrence.
Combination approaches
Exploring the Potential use of Alternate Neoantigens for the Generation of Therapeutic Cancer Vaccines to Improve Efficacy of Immune Checkpoint Blockade- Thamizhanban Manoharan, National University of Singapore, Singapore
To increase the pool of targetable, shared, tumor-specific neoantigens in tumors with a low to moderate tumor mutational burden, such as microsatellite-stable colorectal cancer (MSS CRC), Thamizhanban Manoharan, a graduate student in Gloryn Chia’s lab, turned his attention to circular RNAs. circRNAs are highly stable, closed-ring structures that are formed by “backsplicing” between an upstream 5’ and downstream 3’ splicing site, frequently creating novel “back splice junctions” (BSJ) between included exons. circRNAs are abundantly produced by cells, and their main functions are to control of gene expression (by acting as a microRNA sponge), to regulate protein interactions (by acting as a protein sponge or scaffold), and to act as templates for IRES or m6A modification-dependent translation. Manoharan and colleagues sequenced six paired MSS CRC and normal samples, and used CIRIquant to identify and quantify tumor-associated circRNAs. Once potentially translatable open reading frames were identified with the TransCirc database, the team used pVACbind to predict strong HLA-A*11:01 binders. From approximately 150 upregulated circRNAs in the tumor, the researchers could identify three potentially immunogenic peptides from the BSJ of circMYN9 – a circRNA that was upregulated in 5 out of the 6 tumor samples. Divergent RT-PCR and sequencing was used to verify the presence of these circRNAs in the patient samples, including identification of the BSJ. The immunogenicity of the candidate neoantigens was validated with IFNγ ELISpot, tetramer, and patient-derived organoid killing assays with MYH9 peptide-stimulated healthy donor-derived naive CD8+ T cells. Killing of organoids endogenously presenting the MYH9 peptides was HLA-dependent. circMYH9 was found in the cell-free RNA from plasma samples of two patients tested. Analysis using the exoRbase 2.0 database confirmed high levels of circMYH9 in extracellular vesicles (EVs) in the blood of patients with CRC, versus no detectable levels of circMYH9 in EVs in the blood of healthy donors. Manoharan and his colleagues are now working on identifying the TCR sequence that recognizes circMYH9.
Tiragolumab improves PD-L1 blockade by acting on macrophages and Treg cells- Ruozhen Hu, Genentech, USA
Tiragolumab is a human anti-TIGIT monoclonal antibody currently under active clinical development. Ruozhen Hu presented data on the proposed mechanisms of action of anti-TIGIT antibodies, including direct ligand blockade, depletion of TIGIT-expressing Tregs, or structural modulation of the TCR synapse. More than ten different antibodies are in development, and they mainly differ in the Fc portion (IgG1 [as in tiragolumab], IgG4, Fc-disabled, or Fc-enhanced). To better understand the mechanisms of action of tiragolumab, data from the clinical trial CITYSCAPE were analyzed. In this trial, patients with NSCLC were treated with tiragolumab plus atezolizumab or placebo plus atezolizumab; encouragingly, the former group had a longer median overall survival. Hu and team performed bulk RNAseq on the tumor samples and assessed gene signatures of lymphocytes and stromal cells. The CD8 effector gene signature and, more surprisingly, a tumor-associated macrophage gene signature were both associated with better response to the combination treatment. Therefore, TAMs might be associated with the clinical outcome of anti-TIGIT plus anti-PD-L1 treatment. Comparing serum obtained before and after treatment revealed an increase in myeloid cell-expressed proteins (including CD163, CSF1R, and MARCO, the genes for which were confirmed to be expressed in the myeloid cell RNASeq data) in the combination therapy group, and patients with higher levels of these circulating proteins had longer survival. Hu’s hypothesis for these effects was that tiragolumab leverages tumor myeloid cells via FcγR. To validate this in an experimental system and evaluate the mechanism, Hu and team turned to the mouse CT26 model. The researchers showed that mIgG2a, but not mIgG2b, anti-TIGIT antibodies drive tumor rejection when combined with anti-PD-L1. The IgG2a has more activating FcγR engagement than the IgG2b antibody. The Fc-silent version (LALAPG) was ineffective. Gene expression analysis of TAMs from the mouse tumors showed that Fc-active anti-TIGIT antibodies drove antigen presentation and chemokine gene programs, which were most upregulated with the IgG2a antibody, and further enhanced when combined with anti-PD-L1. In tumoral CD8+ T cells, IgG2a anti-TIGIT reversed the presence of cells with a T cell exhaustion transcriptomic signature, and enhanced those with a memory-like state, while the IgG2b version had the opposite effect. Furthermore, the suppressive capacity of Tregs was reduced in the anti-TIGIT and combination groups, as compared to anti-PD-L1 alone. To determine whether the efficacy of the treatment was dependent on macrophages, these cells were depleted in the model, reducing the shift from exhaustion to memory in the T cells. Therefore, multiple modes of action contribute to the clinical benefit of tiragolumab and atezolizumab, which are associated with effects on myeloid cells, Tregs, and CD8+ T cells, all of which show Fc dependency.
Basic research findings
Targeting pathogenic myelopoiesis in cancer- Miriam Merad, Mount Sinai School of Medicine, USA
Despite the established role of tumor-associated macrophages (TAMs) in the modulation of tumor response to ICB, there are no FDA-approved macrophage-targeted therapies for cancer. Miriam Merad discussed how the development of myeloid-targeted therapies has lagged because macrophages can have both pro- and anti-tumor properties, and the defining features of these different molecular states remain unknown. Therefore, her lab performed extensive single-cell profiling of tumors to search for conserved macrophage molecular programs across human tumors and species. In earlier work, a macrophage molecular cluster dominated by Trem2 transcripts accumulated in most tumors in humans and mice, and this cluster dampened NK cell accumulation and function in lung cancer. Merad then described more recent work demonstrating that an IL-4 signaling module was enriched in macrophages and DCs that infiltrated murine and human NSCLC. IL-4 is a strong inducer of the macrophage repair program, and anti-IL-4 treatment in mice reduces tumor progression of KP (KRASmut tp53-/-) tumors. To better delineate the role of IL-4 in macrophage lineage development, IL-4R was specifically deleted from early myeloid progenitors, which reduced myelopoiesis in the bone marrow and reduced tumor burden in the KP mice. Analysis of the macrophages in the tumors of IL-4R-deleted mice revealed that they were reprogrammed into an immunogenic phenotype, resulting in the accumulation of NK and T cells. The IL-4R-blocking antibody dupilumab is currently being tested to treat R/R NSCLC. Fifteen patients have been included in analyses so far, with partial responses seen in 2 and stable disease seen in 5 patients. Searching next for drivers of pathogenic myelopoiesis, Merad and team performed combined scATAC/RNAseq analyses along the myeloid lineage to assess the genetic cues driving the enhanced myelopoiesis induced by tumors. This revealed a strong induction of a stress response gene program along the myeloid lineage. There was a tumor-driven stepwise increase in the transcription factor Nrf2 and its downstream genes along the myeloid lineage. This stepwise increase correlated with the appearance of more immunosuppressive macrophages (TREM2+) and reduced accessibility of IFN response genes along the myeloid lineage. Myeloid-specific Nrf2 deletion reduced myelopoiesis in the bone marrow and macrophage accumulation in the TME, and slowed tumor progression. Additionally, it resulted in the reprogramming of tumoral macrophages into an immunogenic phenotype that unleashed NK and T cell immunity. When NK cells were deleted in this model, the antitumor effects were lost. Finally, Merad discussed the impact of age, as it is a major risk factor for cancer and is known to enhance myelopoiesis. KP tumor growth was accelerated in the older mice, and transplant experiments of aged bone marrow to young mice and vice versa showed that aged bone marrow was sufficient to enhance tumor growth in young mice, and that young bone marrow transplanted in old mice reduced cancer progression. In the lung tumors of old mice, there were more IL-1α/β-producing myeloid progenitors, and these “old” myeloid cells produced more IL-1α/β in response to tumor debris. Blocking IL-1α/β reduced lung tumor initiation in old animals more potently than blocking IL-1α or IL-1β alone. In conclusion, Merad suggests a two-hit hypothesis: pathogenic myelopoiesis is initiated in the bone marrow, which sets the stage for further dysregulation in the TME. Targeting this process early in the bone marrow might be an effective antitumor strategy.
A stem-like CD4 T cell controls immunity to cancer- Haydn Kissick, Emory University, USA
What defines why patient tumors have such a vastly different degree of T cell infiltration? This is the question Haydn Kissick and his lab have been addressing. Kissick reviewed their past work analyzing the two major CD8+ T cell classes found in human tumors, a “stem-like” CD8+ T cell population (TCF-1+) and an “effector” population expressing one or more inhibitory receptors (PD-1, TIM-3), and often granzyme B (GZMB). Taken out of the tumor, the stem-like cells respond to stimulation with anti-CD3, anti-CD28, and/or IL-2 by proliferating and upregulating GZMB, becoming early effector-like T cells, while the effectors cannot proliferate, but maintain GZMB functionality. Interestingly, differentiation to the effector state appears restricted to the tumor, as such cells can not be found in tumor draining lymph nodes (TDLN), despite presumed priming of these cells in the LN and, in the case of LCMV infection, an abundance of CD8+ effectors in the LN just 1 day after infection. In tumors, antigen-presenting niches can be observed, with myeloid cells (probably dendritic cells) surrounded by both CD4+ and CD8+ stem-like (TCF-1+) cells, likely producing, uniquely in the tumor, the effector cells necessary for tumor control. What drives this two-step process of priming in the LN, but effector differentiation in the tumor for tumor antigens? CD4+ cells in tumor and TDLN appeared similar to each other in gross phenotypes based on analysis of lineage-defining transcription factors, with some Foxp3+ regulatory T cells and a smaller number of EOMES+ cells, but most expressing TCF-1 and lacking other lineage markers (Tbet or BCL6) in the TDLN. Taken out of the tumor and stimulated, the TCF-1+ CD4+ T cells proliferated and differentiated according to what other differentiation factors were included (into Th1 cells with IL-12, into Tregs with TGFβ and IFN, etc.), demonstrating significant plasticity. Dendritic cells from the tumor, without IL-12 added, could similarly drive TCF-1+ CD4+ T cells toward a Th1 phenotype. The already differentiated cells, however, did not proliferate, and were locked in their original state. Switching to a mouse tumor model expressing the well characterized antigen gp66 from LCMV similarly showed that this pattern was replicated for the antigen-specific CD4+ cells (with the exception of more Tregs in the mouse tumors), and that the pattern was established within the first week after tumor implantation. Adoptive transfer of antigen-specific cells from the TDLN to non-tumor-bearing mice, followed by challenge with LCMV, showed that these cells rapidly proliferated and differentiated to Th1-like cells, and thus retained functional potential. Importantly, the presence of Th1 CD4+ cells in the tumors, although rare, may be an important prognostic factor based on a small group of RCC patients with intratumoral Th1 CD4+ cells, who showed dramatic and durable responses to ICB. The TCF-1+ CD4+ T cells were found in the TDLN of almost all patients though, so the major questions are “why don’t these become Th1 cells”, and “when they do, why does this actually help the antitumor immune response?”. Blockade of inhibitory receptors had no effect, but Treg elimination (either with an antibody or in Foxp3-DTR mice) massively shifted the cells toward a Th1 phenotype in the TDLN, and resulted in tumor control, suggesting that Tregs were controlling the “tolerant” vs “effector” programs. Tumor control was dependent on both CD4+ and CD8+ T cells following Treg depletion, with tumor-specific CD8+ T cells massively expanded in the TDLN, just like they were following viral infection. IFNγ appeared to play a significant role in the communication between CD4+ and CD8+ T cells in the TDLN. As an alternative to potentially toxic Treg depletion, Kissick showed that ectopically expressing Tbet in T cells would mimic the Treg depletion effect on Th1 and effector CD8+ T cell stimulation. Kissick closed with an example of a patient with a complete and durable response to anti-CTLA-4 and anti-PD-1 therapy. In contrast to most other patients, this patient showed high levels of Th1 CD4+ cells in the TDLN and tumor, and a high level of effector CD8+ in the TDLN, strongly supporting the results observed in preclinical studies.
PD-1/cDC1 Axis in the Tumor Draining Lymph Node Regulates Stem-like CD8+ T Cell Differentiation- Jyh Liang Hor, National Institutes of Health, USA
The tumor-draining lymph node (TDLN) can serve as a reservoir for stem-like CD8+ progenitors, and Jih Liang Hor used highly multiplex tissue imaging (IBEX) coupled with an optical tissue clearing method (Ce3D) (as described by Ron Germain) to develop 3D images of tumor-draining lymph nodes to investigate how the differentiation process is controlled. This technique can identify the spatial positioning of multiple different cell types, and can assess functional and phenotypic states. A small number (500) of EGFP-labeled OT-I cells were transferred into recipient mice inoculated intradermally with OVA-expressing KP tumor cells, and the TDLN were harvested across several time points for imaging analysis. At day 7, the OT-I cells had massively expanded, and there were three major subpopulations, most of which were TCF-1hi stem-like cells in the T cell zone of the TDLN. A small subset of these stem-like cells expressed high levels of PD-1 and the stem marker SLAMF6. These SLAMF6+ stem-like cells were spatially associated with cross-presenting cDC1s and were uniquely retained within the TDLN. TCF-1- effector cells were on the periphery and presumably exiting the DLN. The high expression of PD-1 on the SLAMF6+ cells suggested some level of TCR signaling, and the researchers hypothesized that some stem-like cells continued to receive antigenic signaling during this late differentiation phase (~10 days beyond the initial priming [the time of tumor implant]). The resolution of the imaging was sufficient to allow discrimination of markers in the nucleus versus the cytoplasm. So, staining of the transcription factor NFAT, which translocates from the cytoplasm into the nucleus upon TCR engagement in lymphocytes, was used to show stained nuclei in cells closely associated with cDC1s. To determine the function of this late antigen presentation, a mouse transgenic model (XCR1 DTR mice) in which cDC1s could be selectively depleted by diphtheria toxin administration was used. Depletion on Day 5/6 after tumor implant, a time point at which robust formation of these clusters could be observed, resulted in a significant reduction of stem-like cells in the TDLN. Therefore, this late antigen presentation phase may be crucial to sustain an ongoing expansion of stem-like cells. Looking into the endogenous polyclonal T cell response to the OVA peptide using LN tissues from mice that did not receive an OT-I transfer, the formation of these PD-1+SLAMF6+ clusters could be detected, but they were not as numerous. The TCR affinity of the stem-like cells was estimated by normalizing their tetramer staining intensity with CD3 or TCR expression. The compartment of cells containing the highest-affinity cells also expressed the highest levels of PD-1, suggesting these high-affinity cells may drive the expansion of stem-like cells. These late antigen presentation niches could function as maintenance hubs where clones are selectively stimulated over time. Interclonal competition based on TCR affinity may result in highest avidity clones receiving the most stimulation. Thus, controlling their expansion at the stem-like state would ensure a simultaneous amplification of both the high-affinity stem-like clones and their effector progenies. The researchers followed the average TCR affinity of both subpopulations in the TDLN, and a steady increase in their average TCR affinity was observed over time. PD-1 from these stem-like cells readily polarized toward the synaptic front between the T cell and the DCs, suggesting that PD-1 molecules were actively engaged with the dendritic cell-presented peptide:MHC during this late antigen presentation phase. PD-1 engagement may prevent these high-affinity stem-like cells from undergoing terminal differentiation. Given the long-term use of anti-PD-1 therapy in patients, the researchers assessed whether the disruption of the PD-1 signaling axis with ICB would dysregulate the formation of these high-affinity stem-like cells. Tumor-bearing mice were treated with anti-PD-L1 and anti-PD-L2 antibodies starting on day 4, and a week later, stem-like cells in the TDLN were assessed. There were fewer stem-like cells, their expression of TCF-1 and SLAMF6 was downregulated, and there was a loss of high-affinity cells. Therefore, these data suggest that PD-1 plays a role in the formation of these high-affinity stem-like cells, and that care should be taken in the duration and regularity of anti-PD-1 therapy.
Identifying CD8+ T Cells Necessary for Anti-melanoma Memory- Marcus Bosenberg, Yale University, USA
After summarizing his work on the role of KDM5B and SETBP1 in upregulation of retroviral elements and the subsequent important RNA-induced type I interferon response and on development of a decoy-resistant IL-18 (insensitive to IL-18 binding protein inhibition) and its activity alone and in combination with ICB in ICB-resistant tumors, Marcus Bosenberg discussed research into a T cell subset expressing IL-7 receptor (IL-7R) with features of stemness and potential effector function. Bosenberg began by developing a series of variants of the GEMM mouse YUMM with mutations induced by irradiation, such that these cells still were not spontaneously rejected when implanted. However, these variants, and additional variants where immunodominant antigens (OVA or gp33) were further added, did respond to ICB, and survivors completely rejected tumor rechallenge, demonstrating the creation of memory that lasted at least 2 years. This memory was CD8+ T cell-dependent. When the S1P inhibitor FTY720 was used to restrain lymphocytes in the lymph node, rechallenge led to tumor outgrowth. This suggests loss of the memory response, and that CD8+ T cells mediating functional anti-melanoma memory resided in lymphoid organs, potentially a source for replenishment of antitumor TILs. The CD8+ T cells residing in the lymph node had a memory and stem-like phenotype, expressing TCF7, IL-7R, and some granzyme B, but a low exhaustion score. The combination of IL-7R blockade during priming, with or without anti-PD-1 and anti-CTLA-4 therapy, resulted in melanoma recurrence, suggesting a compromised memory response was formed. When the researchers used these antigen-specific IL-7Rhi CD8+ T cells for adoptive cell transfer (ACT) before tumor implantation, it protected the mice from tumor outgrowth. This strategy only required 10,000 cells and no lymphodepletion. IL-7R and TCF7 can be upregulated on CD8+ T cells with the drug RG108, which is a non-covalent pan-DNA methylase inhibitor. Ex vivo RG108 pretreatment of IL7Rlo T cells improved their antitumor function after ACT. Finally, confirming these cells were also related to outcomes in patients with melanoma, TCGA data showed that the signature of cells with high IL-7R expression correlated with better outcomes. Therefore, this IL-7Rhi CD8+ T cell population is located in lymph nodes and in the tumor, has stem-like and partial effector features, and is functionally capable of effective ACT responses.
CD40 Agonist Systemically Drives the Differentiation of Tumor-specific and Bystander CD8 T Cells into a Highly Activated PD1+ KLRG1+ Cytotoxic State- Jacqueline Ling Yee, University of California, San Francisco, USA
Anti-CD40 agonists (agCD40) can activate antigen-presenting cells, such as dendritic cells, and drive the expansion of CD8+ T cells. AgCD40s have shown great promise in preclinical models. However, agCD40 treatment of patients, even when used in combination with PD-1 blockade, has delivered rather disappointing results. Jacqueline Yee, a graduate student from Matt Spitzer’s lab, set out to gain a better understanding of the effects agCD40 alone or in combination with PD-1 blockade has on T cells. While anti-PD-1/PD-L1 therapy is known to drive the differentiation of Tpex (PD1+TCF1+TIM3-) cells into a terminally exhausted state, it is not known whether particular T cell subsets are enhanced by agCD40. AgCD40 and anti-PD-L1 monotherapies similarly moderately reduced tumor growth in LLC and MC38 tumor models compared to untreated controls. However, CYTOF analysis 7 days after therapy showed that agCD40 treatment (but not anti-PD-1 treatment) resulted in a massive expansion of PD-1+KLRG1+ T cells in the tumor, spleen, blood, and tumor-draining lymph nodes. Additional studies in a LLC-gp33 model confirmed that these cells contain tumor-specific clones. PD-1+KLRG1+ T cells display an intermediate expression of TOX, CD39, and other exhaustion markers, but despite expression of KLRG1, do not resemble short-lived effector cells. With the confirmed expression of granzyme B and perforin, PD-1+KLRG1+ T cells have strong cytotoxic potential, which still needs to be experimentally verified. Combination of agCD40 and anti-PD-L1 in both the LLC and MC38 models did not confer a synergistic effect compared to the monotherapies in tumor growth control. Notably, the combination of agCD40 and anti-PD-L1 resulted in reduced expansion of PD1+KLRG1+ CD8+ T cells compared to agCD40 monotherapy, suggesting, perhaps, activation-induced cell death (AICD) due to overstimulation.
Chimeric Antigen Receptor T Cells and Carbohydrate-Based Cancer Targets- Avery D. Posey, University of Pennsylvania, USA
Avery Posey discussed various approaches to improve CAR T cell therapies, and started his talk with tumor glycotargets that are generated due to differential glycosylation in tumors. Early termination of O-glycosylation results in cell surface Tn antigen expression which is commonly found on cancers of multiple histologies. A Tn version of the protein mucin 1 (MUC1) was found to be highly expressed on the surface of tumor cells, and has an important immunosuppressive function through recruiting MGL+ M2-skewed macrophages, as well as immature and tolerogenic DC. The exposure of Tn-glycopeptide to MGL+ monocytes triggers IL-10 production and IL-10-producing CD4+ Tregs. To model glycosylation changes in mice, the researchers used the murine KPC pancreatic cancer cell line, and genetically knocked out Cosmc, a T-synthase chaperone protein, allowing buildup of the Tn antigen. The resulting Tn+ mPDAC tumors showed increased growth and lung metastases in vivo, and a change in the distribution of infiltrating myeloid cells, with more M-MDSC and M2 TAMs, and fewer cDC1s. Anti-TnMUC1 CAR T cells eliminated Jurkat T cell leukemia in vivo, but showed no activity against normal human primary cells or in a human MUC1 transgenic mouse. Another glycotarget Posey and team investigated is fibronectin, an extracellular matrix protein that is also expressed on some tumors. Using an antibody recognizing a Tn-containing oncofetal glycoform of fibronectin, fibronectin-targeting CAR T cells were engineered. These CAR T cells killed PC3 cells in vitro and controlled tumor growth in a PC3 xenograft model. Additionally, CAR T cells targeting TnCD19 were shown to specifically target malignant Tn+CD19+ B cells and spare Tn-CD19+ cells, which has potential to prevent B cell aplasia in patients with leukemia and lymphoma treated with CD19-CAR T cells. CD44 is another Tn-modified target that Posey has identified, and the targeting antibody is undergoing optimization. Investigating approaches to improve the cell killing capabilities of CAR T cells, Posey and team found that loss of IFNγR1 expression on tumor cells allows them to evade CAR T cell killing, and CAR T cells lacking IFNγ production lose the ability to lyse tumor cells (as also observed by others). To overcome deficiencies in IFNγ-related killing, Posey found that TLR4 and TLR2/6 agonists could sensitize IFNγR-KO tumor cells to CAR T cell killing. Some clinical trials are now being conducted in which LPS is delivered intratumorally. Posey tested this in mice, and found that it slightly delayed tumor growth in IFNγR1-deficient tumors. Finally, Posey described how they have armored CAR T cells with IL-12 or IL-23 for enhanced antitumor activity against low-antigen expressing tumors. When such CAR T cells are used against the IFNγ-resistant PC3 cells, there is antitumor activity. However, engineering these CAR T cells to secrete IL-12 results in differentiation during the manufacturing. Therefore, the researchers have developed immunocytokines comprising an antibody conjugated to IL-12, IL-18, or IL-23 targeting the invariant linker regions among currently approved CARs. These immunocytokines can be added post-manufacturing and, importantly, allow for titration of the cytokines. In vivo testing showed increased efficacy of the CD19 CAR T cells with the addition of immunocytokines in the NALM6 model, resulting in better survival and persistence of the CAR T cells.
Improving antitumor immunity
DaNGeRous Shaping of immunity to Cancer- Jonathan Lim, Francis Crick Institute, UK
Type 1 conventional dendritic cells (cDC1s) are indispensable for the control or rejection of immunogenic tumors, but also for successful immune checkpoint blockade and adoptive T cell therapy. Jonathan Lim discussed TCGA data that showed that high expression of the cDC1 signature correlates to better overall survival (OS) in patients with several subtypes of cancer. One of the top genes in the cDC1 signature is DNGR-1 (CLEC9A), which has been found to predict better OS in various tumor types. The ligand for DNGR-1 is F-actin, and receptor signaling promotes phagosomal rupture, permitting cross-presentation of antigens from dead cells to CD8+ T cells. Given its expression in both human and mouse cDC1s, Lim was interested in experimentally examining the functional role of DNGR-1. DNGR-1 was knocked out, and tumors were induced with MCA (3-methylcholanthrene) carcinogenesis. Just as in Rag1-/- mice lacking all T cells, or Batf3-/- mice lacking cDC1s, DNGR-1-deficient mice were more susceptible to tumor development compared to WT controls. Tumor cell lines from MCA-induced primary tumors in WT, RAG1-/-, Batf3-/-. and Clec9Acre/cre or Clec9Agfp/gfp were then established (as Bob Schreiber had done to demonstrate immunoediting of neoantigens) to collect a library of primary cell lines for further analysis. These cell lines were injected into WT recipient mice to compare tumor growth. Primary sarcoma lines derived from DNGR-1-deficient hosts were highly immunogenic, similar to cell lines grown in immunodeficient mice, suggesting lack of immunoediting. These results were confirmed in a model of colon cancer in which DNGR-1-deficient mice were also shown to be more susceptible to tumor development compared to WT controls. The hypothesis Lim and colleagues developed for immunoediting based on these results was that in DNGR-1+ (WT) mice, DNGR-1 binds to F-actin and consequently captures the repertoire of 80 to 90 F-actin binding proteins (FABP), selectively presenting FABP neoantigens and generating an immune response, which ultimately selects for cells not expressing these FABP neoantigens (immunoedited tumor cells). In the knockout setting, the new antigen that is decorating the ligand itself might not be edited out. To test this hypothesis, the researchers performed whole-exome sequencing and neoantigen prediction on the regressor and (immunoedited) progressor cell lines. This revealed that although there was no overall difference in the number of predicted neoantigens between these cell lines, neoantigens present in FABP were enriched in immunoedited progressor cell lines, supporting a direct role for DNGR-1 in cancer immunoediting.
Catalytically Inactive cGAS Potentiates Antitumor Immunity- Alexander Keller, EPFL, Switzerland
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway mediates sensing and regulation of the cellular response towards double-stranded DNA from a variety of different sources and has gained a lot of attention in recent years, specifically related to antitumor immunity. Alexander Keller and his colleagues in Andrea Ablasser’s lab, introduced a mutation in cGAS (D307N), in order to render cGAS catalytically inactive to study the STING-independent impact of cGAS on antitumor immunity (such as a role of cGAS in genomic instability). While the growth of CT26 tumors with cGAS KO could no longer be controlled in mice, and re-introduction of WT cGAS fully prevented tumor growth, introduction of D307N cCAS controlled the tumor to some extent, but not fully. Further analysis demonstrated increased T cell infiltration and activation in D307N cCAS compared to cGAS KO CT26 tumors, and confirmed CD8+ T cell-dependent tumor control in mice bearing D307N cGAS CT26 tumors. Gene set enrichment analysis revealed hallmarks of genomic instability (e.g., activation of DNA repair and G to M checkpoint), leading to a de novo increased load of high to moderate impact variants, and a shift towards a more clonal state of these tumor variants. To confirm that this effect was indeed due to D307N cGas expression, D307N cGAS CT26 cells were kept in culture for about half a year. For these cells, an expansion of variants and other hallmarks of genomic instability – such as increased micronuclei, delayed entry into S-phase, and increase 53BP1 foci – were observed over time. This led to the hypothesis that the observed variants will produce neoantigens that can stimulate a robust immune response. In a first step to test this hypothesis, Keller observed an increase in productive T cell clones and synergy with anti-PD-1 treatment. The researchers also tested other models, and were able to recapture their observations in the E0771 breast cancer model, but not in “colder” tumor models (MC38, B16), potentially related to the immunogenicity of the cancer model used, although this needs further follow-up. Of note, only the DNA binding capacity of cGAS, but not its dimerization or nucleosome binding ability, had an impact on tumor growth.
The Exonuclease TREX1 Constitutes an Innate Immune Checkpoint Limiting cGAS/STING-Mediated Anti-tumor Immunity- Klaus Heger, Genentech, Inc, USA
Activation of the cGAS/STING pathway in microbial infections contributes significantly to T cell activation and expansion, but activation of this pathway in tumors is limited. Type I interferons are key signaling molecules induced by this pathway, but toxicity with direct interferon drugs has required better delivery approaches, which, to date, are still under development. Klaus Heger investigated the concept of inhibiting negative regulators of the cGAS/STING pathway as an alternative approach to increase its activation in cancer. TREX1 is a cytoplasmic exonuclease that degrades single- and double-stranded DNA, removing the stimulatory nucleic acids, and thus reducing cGAS/STING pathway activation, with supporting data in humans (inherited loss leading to various interferonopathies) and in murine models (knockout leading to inflammation). Furthermore, there is data to suggest that tumors are dependent on TREX1 suppression of this pathway. Heger and colleagues began by knocking out TREX1 in various murine tumor cell lines, and observed induction of particular interferon-stimulated genes (ZP1, CCL5, and CXCL10), with some variability between cell lines. CT26 was chosen for further investigation as an inflamed tumor model that also showed good induction of the marker genes. As expected, in NSG mice, there was no impact of TREX1 KO, but in immunocompetent Balb/c mice, KO delayed tumor growth, extending survival and occasionally led to complete responses. An antibody to the Type I IFN receptor IFNAR1 abrogated the improved tumor control, indicating it was IFN-dependent, and knockout of cGAS abrogated tumor control, suggesting the importance of the cGAS/STING pathway in this response. scRNASeq analysis of tumor samples from TREX1 KO mice demonstrated a decrease in immunosuppressive macrophage subsets and an increase in inflammatory monocytes, indicating a less suppressive tumor microenvironment. The largest effect was observed on CD8+ T cells. Exhausted and precursor-exhausted cells were significantly reduced, and cytotoxic subsets were increased (CCL5+ populations and populations expressing an IFN-activated signature), with specific upregulation of cytotoxic markers, such as granzyme B and SCA-I. Combining these TREX1 KO effects with anti-PD-1 therapy led to significant synergy, curing most animals, whereas either treatment alone yielded few complete responses. Altogether, this work validated the concept of targeting negative regulators of this immune stimulating pathway to enhance cancer cell-intrinsic immunity.
Overlapping and Distinct Mechanisms of Effective Neoantigen Cancer Vaccines and Immune Checkpoint Therapy- Matthew Gubin, MD Anderson, USA
To be able to develop more effective cancer vaccine-based combination therapies, Matt Gubin set out to study, in a controlled model system, the mechanism of how efficacious neoantigen vaccines and immune checkpoint blockade (ICB) enhance antitumor immunity. Gubin and his team used Y1.7LI, the immunotherapy-resistant BrafV600EPten-/-Cdkn2a-/- YUMM1.7 melanoma cell line from Marcus Bosenberg, modified to express both the mutated MHC class I neoantigen mLama4 and the mutated MHC class II neoantigen mItgb1, as a model system. In this model, treatment starting at day 7 post-tumor transplant with ICB (either anti-CTLA-4, anti-PD-1, or anti-CTLA-4/anti-PD-1) or with a synthetic long-peptide vaccine targeting mLama4 plus polyI:C resulted in similar rates of tumor rejection. A control vaccine containing polyI:C and an irrelevant peptide antigen was ineffective. Rejection was CD4+ and CD8+ T cell-dependent. Longitudinal studies demonstrated that both ICB and vaccination resulted in peak T cell infiltration at about day 15, and that the neoantigen vaccine induced the greatest expansion of mLama4-specific CD8+ T cells. scRNAseq and scTCRseq revealed that mLama4-specific vaccination increased neoantigen-specific PD-1+TCF-1+ plastic/stem-like CD8+ T cells and proliferating CD8+ T cells, while anti-PD-1 treatment with or without anti-CTLA-4 induced the expansion of a familiar exhausted/activated PD-1+TCF-1-Tox+ CD8+ T cell cluster. The combination ICB (anti-CTLA-4/anti-PD-1) strongly induced a cluster of PD-1+TCF-1-Bhlhe40hi highly activated neoantigen-specific CD8+ T cells. Interestingly, the Bhlhe40hi gene signature correlated with increased survival probability in patients with various cancer types after treatment with anti-CTLA-4 and/or anti-PD-1/PD-L1. CD8+ T cell-specific knockout of the transcription factor Bhlhe40 showed that it was required in CD8+ T cells for anti-PD-1 antitumor responses1, but was dispensable in CD8+ T cells for anti-CTLA-4 efficacy. In contrast to mLama4-specific vaccination or anti-PD-1 monotherapy, anti-CTLA-4 or anti-CTLA-4/anti-PD-1 treatment induced ICOS+Bhlhe40+IFNγ+ Th1-like CD4+ T cells. To study whether Bhlhe40 was required for the function of ICOS+ Th1-like CD4+ T cells, Gubin and his colleagues treated Bhlhe40KO CD4+ T cells with anti-CTLA-4, which led to an increase in ICOS+Foxp3- CD4+ T cells. However, intracellular cytokine staining revealed a shift from IFNγ+ to IL-10+ CD4+ T cells in the absence of Bhlhe40. Additional treatment with an anti-IL-10R antibody partially restored anti-CTLA-4-mediated tumor regression in tumor-bearing Bhlhe40KO mice. Gubin and team next took a closer look at the intratumoral macrophage compartment. In mice treated with a control antibody, 31% of macrophages were CX3CR1+CD206+ M2-like macrophages. In mice treated with immune checkpoint blockade (ICB), this percentage was lower, while the percentage of iNOS+ M1-like macrophages was increased. In contrast, both mLama4-specific vaccination and control vaccination (also containing polyI:C) increased the percentage of CX3CR1+CD206+ M2-like macrophages (also expressing TREM2) as well as iNOS+ M1-like macrophages. When vaccination was initiated later, a greater number of CX3CR1+CD206+TREM2+ M2-like macrophages were observed, and the efficacy of the vaccine was reduced. A TREM2-blocking antibody rescued tumor regression in the majority of mice treated with late vaccination.
Methods to analyze tumor immunity
Gaining Insight into Tumor Immunity by Combining New Highly Multiplex 2D and 3D Imaging with Analytical Tools- Ronald N. Germain, NIAID, National Institutes of Health, USA
With an underlying recognition that immune reactions occur in complex tissues, Ronald Germain discussed multiplex imaging methods, and what information can be gathered from these methods. A 12-plex imaging panel was used to evaluate core biopsies from patients with mesothelioma participating in a Phase II study testing the combination of an anti-mesothelin immunotoxin (LMB-100) and pembrolizumab. This showed specific locations within the tumors (“starbursts”) where an immune response was taking place. The ubiquitous presence of CD4+ T cells in these clusters suggested they were essential for this response. Additionally, some older experiments had shown time-dependent (as rapidly as 2 hours) increases in PD-1 on effector cells following antigen engagement, that cytokine production from CD4+ T cells stopped as soon as they regained motility, and that PD-L1 blockade delayed the reduction in cytokine production. These data suggest a model supporting the importance of both CD4+ and CD8+ T cells and anti-PD-1 therapy to a local IFNγ circuit, stimulated first by CD4+ T cells, in these spatially restricted small starburst clusters, but tuned by upregulated PD-1. This model was consistent with data from Bob Schreiber on the importance of MHC-I and MHC-II antigens being present in the same cell for effective tumor control, and suggests that a solution to ineffective anti-PD-1 therapy will be iterative rounds of infiltration by additional effector cells. Germain then discussed research into the effects of immunotherapy on Tregs in a pancreatic cancer model. Individual cells from a pancreatic tumor were cloned to create phenotypically stable clones, of which, some would grow into immune “hot” and others into “cold” tumors. The hot tumors responded well to combination therapy with anti-PD-1, non-Treg depleting anti-CTLA-4, and agonistic anti-CD40. Imaging showed reduced levels of Foxp3+ Tregs after therapy. Foxp3 lineage tracing showed that therapy did not induce Treg death, but a loss of Foxp3 expression in those cells (ex-Tregs). This effect was restricted to the tumor microenvironment, and was dependent on IL-12 and IFNγ. The ex-Tregs became Tbet+ effector Th1 cells that produced IFNγ and were in close contact to cDC1s. High-resolution imaging of NFAT showed that ex-Tregs had the highest proportion of cells with nuclear NFAT expression, a marker of TCR signaling. Therefore, the Tregs that were being converted with the therapy were among those with the greatest functional TCR engagement, and would be highly suppressive if not converted. Finally, Germain described some incredible work with high-plex imaging by confocal microscopy using an open-source technique called iterative bleaching extends multiplexity (IBEX), with readily available reagents. Up to 8 parameters (up to 12-15 with special equipment) can be used per cycle. Combining IBEX with a clearing method (Ce3D) allows determination of cell interactions, signals, and gradients across tissue. An algorithm (spatial analysis of cell ensembles (SPACE)) was developed to detect and display these patterns. Using this analysis on mouse lung tissue revealed a new type of lymphoid tissue – an immune cluster located near nerve junctions.
The shape of things- Implicit order drives knowledge transfer- Garry P. Nolan, Stanford University, USA
Given that more and more laboratories have complex imaging capabilities, Gary Nolan discussed ways the complex data can be analyzed and combined using various algorithms. One issue is data merging from different analytical techniques (RNA, protein, ATAC, etc.). Nolan explained that thinking about RNA, protein, and epigenome data as languages, and that languages have similar structures in high-dimensional space, allows development of algorithms to merge that disparate data, without having exact bridging information. In an example, Nolan described work in which different labs had performed IBEX imaging, RNA, and ATAC analyses on different regions of the intestines from various donors. It is also possible to merge data from the literature, as shown with an example of merging transcription factor gene expression and in an example of analyzing tonsil tissue stained for surface markers, showing that the cells can be segmented based on phenotypic information from literature. Overlaying these data with RNA data can show gene enrichment in certain areas, without having assessed the RNA directly in the tissue. Nolan and team also developed an approach for tissue schematics, to build an interaction map to assess the interface of various areas of tissue, and to predict and understand what signals generated by local processes modulate the expression of functional markers. The mechanistic information can be confirmed with knowledge from the literature, but this is an incredibly resource-intensive undertaking. To automate this analysis, the Nolan lab uses a large language model (CellFormatica), in which all PubMed abstracts were included (the next version will have all of PubMed in it) to provide literature-based context and rationale. An experimental approach has been set up with 200-300 question/answer formats that the model needs to go through to extract the information from any paper. The researcher provides a gene list and a context to target the search, and asks a question on the role of these genes in that context. The model generates an effect score that provides a quantitative measure of how a certain gene modification impacts a particular hypothesis. For each gene, it provides its role, literature sources, and confidence level based on information available. The language model can then provide information on function and data from the literature, hypotheses for modes of action, and information on the required validation experiments to confirm this hypothesis, all based on the available literature.
By Ute Burkhardt, Ed Fritsch, Maartje Wouters, and Lauren Hitchings
(This Keystone Symposia conference is available for On Demand viewing! Find out more here).



