Can checkpoint blockade benefit patients whose cancers have low tumor mutational burden? To address this question, Subudhi et al. conducted a phase II clinical trial of patients with advanced prostate cancer – a cancer known to have a relatively low mutational load – who were treated with ipilimumab, and evaluated their treatment-induced immune responses along with clinical outcomes. The results were recently published in Science Translational Medicine.
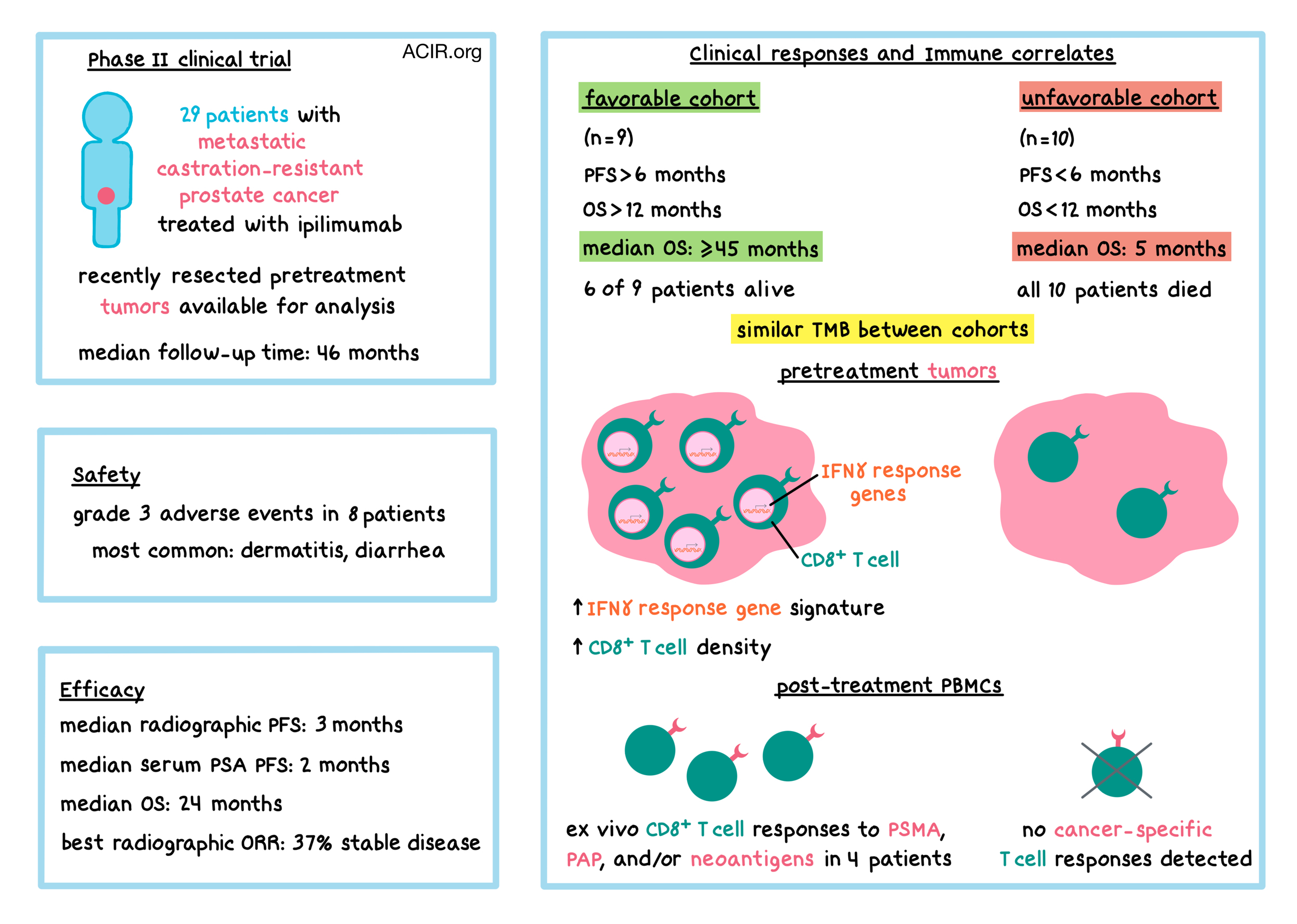
Prostate cancers, which typically respond poorly to immune checkpoint blockade (ICB), generally have a relatively low frequency of mutations and neoantigens compared with cancers like melanoma and non-small cell lung cancer, which tend to respond well to ICB. Nevertheless, a subset of patients with metastatic castration-resistant prostate cancer (mCRPC) may experience clinical benefit from ICB. To test the hypothesis that neoantigen-specific T cell responses may be induced by ICB in some patients with prostate cancer, 30 patients with mCRPC were enrolled in a phase II clinical trial to evaluate clinical outcomes and immune correlates following ipilimumab (anti-CTLA-4) therapy. 29 of the patients received at least one dose of ipilimumab. Patients had to have their prostate cancer resected within 3 months prior to entering the study, and the resected tumor masses were analyzed for mutational load and predicted neoantigens using whole-exome sequencing and RNAseq. Patients received ipilimumab once every 3 weeks for a maximum of 4 doses, and systemic antitumor T cell responses were evaluated after each dose.
At a median follow-up of 46 months from the administration of the first dose, safety and efficacy were evaluated. Eight patients experienced grade 3 adverse events, and no grade 4 or 5 events occurred. The most common grade 3 immune-related adverse events were dermatitis and diarrhea. Progression-free survival (PFS) was evaluated in two ways: radiographic PFS and clinical PFS based on the change in levels of serum prostate-specific antigen (PSA). Median radiographic PFS was 3 months, while median clinical PFS was 2 months. Median overall survival (OS) was 24 months, and 22 patients (76%) died from their disease. 37% of patients experienced stable disease, which was the best overall radiographic response in this study.
To better characterize the subset of patients who may benefit from ipilimumab, Subudhi et al. stratified the patients by clinical outcomes. Two of the 29 treated patients withdrew their consent after starting treatment, and the remaining 27 patients were stratified by their radiographic and/or clinical PFS (rcPFS, whichever occurred first). Nine patients had rcPFS greater than 6 months, and all of these patients had OS greater than 12 months – these 9 patients constituted the “favorable” cohort. The other 18 patients had rcPFS less than 6 months; among these patients, 8 had OS greater than 12 months and 10 had OS less than 12 months. These latter 10 patients constituted the “unfavorable” cohort. The difference in survival between the favorable and unfavorable cohorts was striking: median OS for the favorable cohort was at least 45 months (or greater), compared with 5 months for the unfavorable cohort. Six of nine patients in the favorable cohort were alive at the time of analysis, while all ten patients in the unfavorable cohort died from their disease.
RNAseq of pretreatment tumors showed that IFNγ response pathway and T cell (including cytotoxic and memory T cells) gene signatures were higher in patients within the favorable cohort compared to patients within the unfavorable cohort. Immunohistochemistry staining confirmed increased intratumoral CD8+ T cell density, cytotoxicity (e.g., granzyme B), and activation (e.g., PD-1). These results suggest that tumor-infiltrating lymphocyte density and IFNγ response gene signature may be predictors of response to ipilimumab and may act as prognostic markers of OS.
Looking for potential neoantigens that may have correlated with responses to ipilimumab, the researchers first performed whole-exome sequencing (WES) on resected pretreatment tumors and on matched peripheral blood mononuclear cells (PBMCs) to uncover tumor-specific mutations. The median number of nonsynonymous somatic mutations was 76, consistent with other data for prostate cancer. Interestingly, tumor mutational burden (TMB) was similar between favorable and unfavorable cohorts, and did not correlate with clinical response to ipilimumab.
Next, the researchers used RNAseq to determine which mutations were expressed, and ELISpot with PBMCs to figure out which expressed mutations led to T cell responses. T cell responses against prostate cancer-associated antigens, such as prostate-specific membrane antigen (PSMA) and prostatic acid phosphatase (PAP) were also evaluated. 17 patients were evaluable, including 8 in the favorable cohort and 4 in the unfavorable cohort. T cell responses to PSMA, PAP, and/or neoantigens were found only in four patients, all within the favorable cohort. Responses to three neoantigens identified in two patients (1 neoantigen in one patient, 2 in the other) could not be detected in pretreatment PBMCs, but were detectable and enhanced after treatment. Responses were mutation-specific and appeared to be driven by CD8+ T cells, suggesting that the neoantigens were MHC-I-restricted. The neoantigens were clonal and found in all tumor cells of each corresponding patient. Additionally, all three neoantigens were also predicted by NetMHCpan.
While TMB did not correlate with clinical outcome after treatment with ipilimumab, patients in the favorable cohort had increased intratumoral CD8+ T cell density, an enhanced IFNγ response gene signature, detectable cancer-associated antigen- or neoantigen-specific T cell responses, or some combination of these factors. The results of this study suggest that these factors may identify patients with mCRPC who may benefit from immune checkpoint blockade, and support further exploration of immunotherapies (particularly combinations with other checkpoint inhibitors or neoantigen vaccines) in patients with metastatic prostate cancer.
by Anna Scherer



