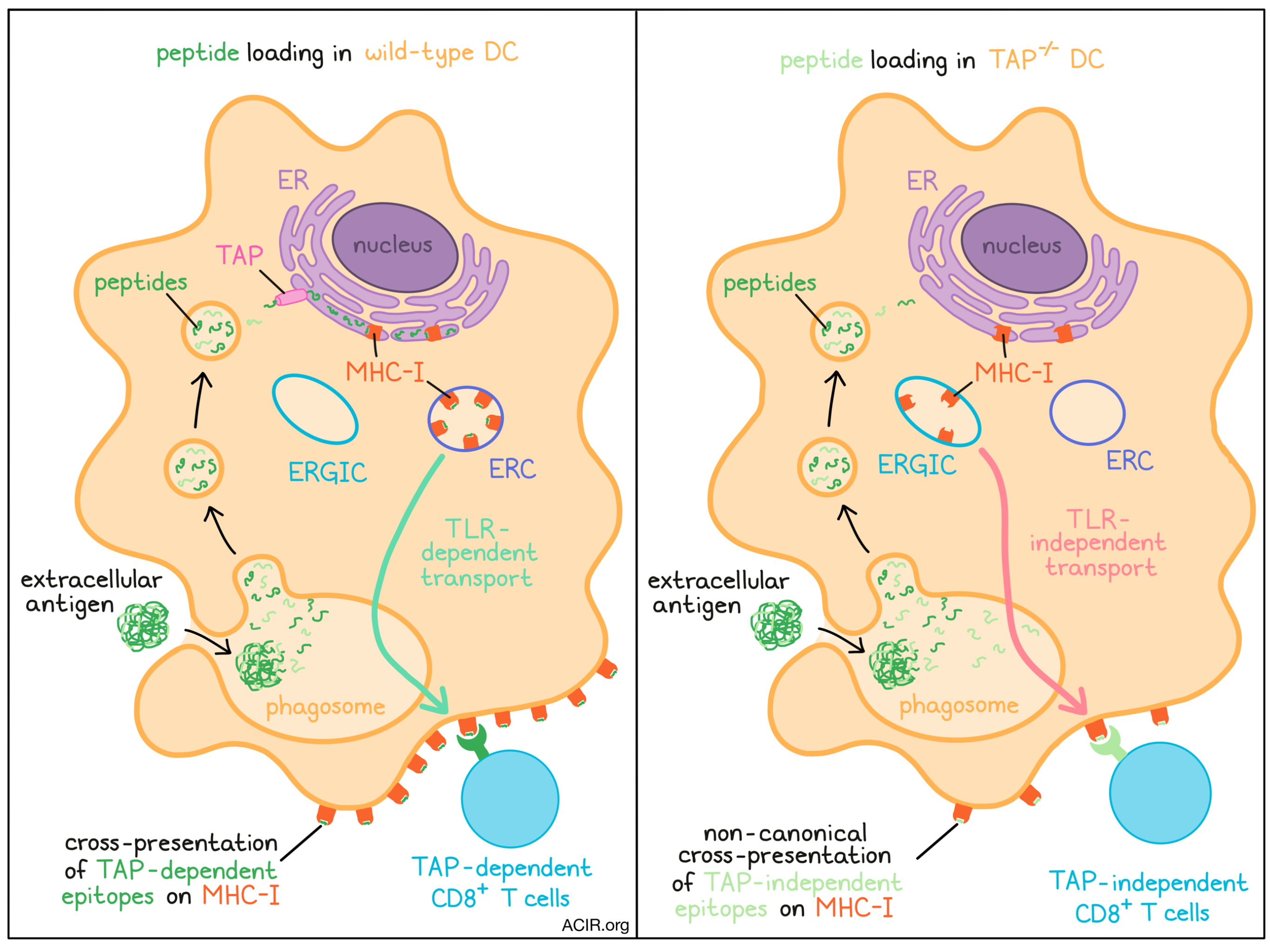
Our classic understanding of antigen presentation to CD8+ T cells involves transport of cytosolic peptides into the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP), where they can be loaded onto MHC class I molecules through the canonical peptide loading complex. Cross-presentation occurs when extracellular antigens are presented on MHC-I, and is critical in priming CD8+ T cell responses against cancer. However, many cancers downregulate TAP, resulting in the expression of alternative (TAP-independent) epitopes which may not be recognized by a T cell response primed by DC’s that contain TAP. Recently reported in Nature Immunology, Barbet and Nair-Gupta et al. investigated antigen presentation and CD8+ T cell responses in the context of TAP deficiency, elucidating an alternative mechanism for cross-presentation.
The researchers began by characterizing the effects of TAP deficiency on MHC-I localization. In wild-type (WT) bone marrow-derived mouse DCs, MHC-I molecules colocalized with markers (Rab11a and VAMP-3) of the endosomal recycling compartment (ERC) and not with markers (ERGIC-53) of the ER-Golgi intermediate compartment (ERGIC). Interestingly, the opposite was observed in Tap1-/- DCs; MHC-I appeared at the ERGIC rather than the ERC, while also declining on the cell surface. Blocking ER-Golgi protein transport in WT DCs with Brefeldin A also altered MHC-I localization in the same manner. Similarly, in human monocyte-derived DCs (moDCs), MHC-I again colocalized with the ERC, but redistributed to the ERGIC when the moDCs were infected with a TAP-inhibiting human cytomegalovirus (HCMV).
Barbet and Nair-Gupta et al. next considered whether the differential MHC-I localization observed with TAP deficiency could affect DC cross-presentation to CD8+ T cells. To investigate, the researchers treated DCs with US6, an HCMV-derived TAP inhibitor, for a short period (2 hours) during which time MHC-I localization was not altered, but classical MHC-I presentation was inhibited. US6-treated DCs could still cross-present extracellular antigen, quantified by proliferation of co-cultured antigen-specific CD8+ T cells. This process was dependent on cathepsin S, a lysosomal protease. Despite their altered MHC-I localization, Tap1-/- DCs could similarly cross-present. These findings indicated that DCs deficient in TAP, either in the short-term (US6 treatment) or long-term (TAP knockout), could reliably cross-present antigen to CD8+ T cells regardless of the intracellular location of MHC-I.
As MHC-I transport from the ERC to phagosomes is regulated by TLR signaling, the authors next hypothesized that the redistribution of MHC-I to the ERGIC might bypass this limitation. The researchers exposed WT or Tap1-/- DCs to extracellular OVA antigen with or without lipopolysaccharide (LPS) and assessed cross-presentation via OT-1 proliferation in coculture. As expected, the ability of WT DCs to cross-present OVA increased with LPS, consistent with the idea that TLR signaling regulates MHC-I ERC–phagosome transport, and MHC-I molecules were observed specifically in LPS-containing phagosomes. Strikingly, Tap1-/- DCs could cross-present OVA just as effectively with or without LPS, and MHC-I molecules were found on phagosomes irrespective of LPS content. The team found Rab11a (an ERC marker) in LPS-containing phagosomes in Tap1-/- DCs, confirming that ERC–phagosome transport still worked - but MHC-I no longer remained within the ERC to be transported by this mechanism. Probing more directly, the researchers found that shRNA-mediated disruption of Rab11a but not Sec22b (an ERGIC marker) blocked phagosome delivery of MHC-I to the cell surface in WT DCs, while the opposite was observed in Tap1-/- DCs.
Additional studies confirmed these findings. In DCs deficient in TLR signaling (Trif-/-Myd88-/-), treatment with Brefeldin A (which blocks protein transport from the ER to the Golgi and so mimics the effect of TAP loss) rescued cross-presentation through this alternative pathway. Similarly, blockade of IKK2, a regulator of ERC–phagosome transport, disrupted MHC-I delivery to phagosomes and cross-presentation in WT, but not Tap1-/- DCs. Overall, these results suggest that the relocalization of MHC-I from the ERC to the ERGIC switches to TLR-independent transport of MHC-I from the ERGIC to phagosomes, where TAP-independent peptides can be loaded. The authors termed this ‘non-canonical’ cross-presentation to distinguish this mechanism from canonical cross-presentation involving TAP-mediated peptide transport to the ER, and passage of TAP-dependent peptide-loaded MHC complexes from ERCs to phagosomes. The authors suggest that this process could potentially risk autoimmunity through presentation of self-epitopes without TLR stimulation; indeed, while virus-infected WT DCs could not cross-present apoptotic cell antigen, Tap1-/- DCs could, and upregulated co-stimulatory molecules (CD40 and CD86), prompting T cell proliferation against OVA expressed on the apoptotic cells.
The authors next tested their findings in an in vivo model of viral infection with hematopoietic TAP deficiency. Irradiated mice were transplanted with wild-type or Tap1-/- bone marrow, infected with a sublethal influenza A dose, and five weeks later, challenged with a lethal dose from a heterotypic strain, eliminating antibody-based control differences. As expected, initial CD8+ T cell responses against TAP-dependent viral epitopes were significantly reduced in the Tap1-/- bone marrow chimeras compared to the controls, but surprisingly, no differences were noted in lung viral titers or mouse weights. After the second, lethal challenge, lung CD8+ T cell responses against TAP-dependent epitopes were again diminished in the Tap1-/- chimeras, yet mice of both groups survived equally well, and evidence of CD8+ T cell activation and antigen experience was observed between groups. Importantly, CD8+ T cell depletion reduced survival of both WT and Tap1-/- chimeras equivalently, revealing that mice with hematopoietic TAP deficiency could still effectively resist disease through a TAP-independent CD8+ T cell mechanism.
To probe the specificity of these TAP-independent responses, the researchers isolated CD8+ T cells from the lungs and spleens of the chimeric mice for coculture with virus-infected WT or Tap1-/- DCs. Interestingly, T cells from WT chimeras produced IFNγ only in response to WT DCs, whereas T cells from Tap1-/- chimeras responded to both Tap1-/- and WT DCs, showing a specificity for TAP-independent epitopes, and suggesting that WT DCs may also present TAP-independent epitopes, although presumably to a lesser extent.
In summary, Barbet and Nair-Gupta et al. demonstrated an alternative pathway for CD8+ T cell cross-presentation by dendritic cells in the absence of TAP functionality. In this context, MHC-I molecules relocate from the ERC to the ERGIC and are delivered to phagosomes independent of TLR signaling. These findings have implications for utilizing immunotherapy against cancer, which can downregulate TAP and present non-canonical peptides. Therapeutic approaches that target TAP-independent CD8+ T cell responses may avoid cancer immune evasion and broaden the arsenal of cancer treatment.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, first co-author Gaetan Barbet and lead author Julie Magarian Blander answered our questions.

What prompted you to tackle this research question?
GB: When the talented Ph.D. student of the lab, Priyanka Nair-Gupta, who just published a Cell paper on cross-presentation, showed in lab meeting new, intriguing data concerning a relocation of MHC-I molecules in absence of TAP, I knew it could lead to a major discovery relevant for vaccines, and antiviral and antitumor immunity. When Dr. Nair-Gupta graduated and left the laboratory for a new position, I had the opportunity to develop the project further, since I have always been fascinated by the recognition of self and non-self by the immune system and its regulation. The opportunity to collaborate with world-renowned virologists at the Icahn School of Medicine at Mount Sinai to develop a mouse model to corroborate our in vitro findings was an exciting challenge and a unique experience.
JMB: It was very much unplanned. Priyanka Nair-Gupta, first co-author on the paper, was a graduate student in my lab studying the regulation of cross-presentation by Toll-like receptors (TLRs). Cross-presentation allows dendritic cells (DCs) to load their MHC-I molecules with peptides derived from internalized microbes or dying cells. Priyanka had found that dendritic cells (DCs) contained an intracellular depot of major histocompatibility complex class I (MHC-I) molecules in endosomal recycling compartments (ERC), and that TLRs augmented cross-presentation by deploying ERC stores of MHC-I to sites of internalized microbial antigen to meet the increased demand for cross-presentation during infection. As follow up to this work, Priyanka and I were merely looking for a way to deplete MHC-I in the ERC, reasoning that cutting off the supply of MHC-I molecules from the ERC would shut down cross-presentation and cripple the CD8+ T cell response to infection. Thinking of ways in which we could drain the ERC stores of MHC-I, we turned to the transporter associated with antigen processing (TAP), which shuttles cytosolic proteasome-generated peptides into the endoplasmic reticulum (ER) for loading onto nascent MHC-I molecules. Without TAP, the supply of peptides for MHC-I molecules falls short, and as a result, smaller numbers of MHC-I leave the ER and reach the plasma membrane. Although we were not sure that the plasma membrane was the source of MHC-I in the ERC, we figured it was worth looking at DCs from genetically engineered TAP-deficient mice and examined their ERC stores and cross-presentation abilities.
What was the most surprising finding of this study for you?
GB: Viruses develop strategies to evade immune responses. It is very rare to uncover mechanisms that counteract those viral strategies. The idea that the pathway that rescues cross-presentation bypasses the control of the Toll-like receptor opens many questions on the regulation of antigen presentation, lymphocyte activation, and the T cell repertoire. This particular context of antigen presentation could also help us to understand how tumor cells evade the immune system, because like viruses, they interfere with the same pathways. This work is not only relevant in the context of immune responses to viral infections, but goes far beyond.
JMB: We had two surprises. We certainly did not find the answer we were initially looking for, and at first we thought we were on the wrong track. We were happy to see that the ERC stores of MHC-I were depleted in TAP-deficient DCs as we predicted, but surprisingly, this did not affect cross-presentation. If anything, TAP-deficient DCs were doing a better job at cross-presentation than wild-type DCs and they did not even need TLR signals to do so. TAP deficient DCs cross-presented antigens irrespective of whether they were derived from microbes or apoptotic cells (self). Baffled, we looked inside TAP-deficient DCs and found that the MHC-I molecules had accumulated in the ER-Golgi intermediate compartment. Was it possible that they could still meet up with internalized antigens and cross-present? We designed a series of experiments to test this and found that ERGIC-accumulated MHC-I molecules used a different pathway of vesicular traffic to reach internalized antigens. This pathway of cross-presentation, which we call noncanonical cross-presentation, relied on the ER SNARE Sec22b and was not controlled by TLRs, explaining the indiscriminate cross-presentation of self and non-self antigens by TAP-deficient DCs. Our findings defined a cell-intrinsic strategy to counter TAP blockade in DCs and rescue CD8+ T cell cross-priming.
The second surprise came when we found that a CD8+ T cell response could be mobilized during viral infection in the absence of TAP expression in hematopoietic cells, and that it even protected the mice against challenge with a lethal dose of virus. These results went against the paradigm that a CD8+ T cell response is severely impaired in the absence of TAP function in hematopoietic DCs. Many clinically important viruses block TAP to evade CD8+ T cells, but some human cancers also downmodulate TAP expression for the same reasons to escape detection by cytotoxic CD8+ T cells. Our findings point to a TAP-independent CD8+ T cell response that could be mobilized under conditions of immune evasion. The field has been preoccupied with studying mostly TAP-dependent CD8+ T cell responses and the need to invoke TAP-sufficient DCs to cross-prime CD8+ T cell immunity. However, a TAP-dependent CD8+ T cell response is not going to be effective in eliminating virus-infected cells and tumors that have disabled TAP. We expect that TAP-dependent CD8+ T cells would get to the tissue and not be able to find their cognate epitopes. Interrogating the nature and specificity of the TAP-independent CD8+ T cell response should open avenues for mobilizing CD8+ T cell responses that are best matched against immune-evasive viruses and tumors.
What was the coolest thing you’ve learned (about) recently outside of work?
GB: The global pandemic affected research dramatically. However, I was proud to be part of a scientific community that could overcome the travel and networking restrictions not only to keep alive productive research programs, but also to reach out to non-scientific audiences that needed reliable information. Of course, this crisis highlighted the gap between the scientific community and the public, but I am very optimistic it will overall lead to a better and more direct communication between scientists and the public.
JMB: This is a difficult question to answer when the past year has been dominated by the COVID-19 pandemic. Travel, hobbies, normal life have all been suspended. I consider myself lucky to be able to continue working when so many have lost their jobs and primary sources of income. On a bright note, I am getting my COVID-19 vaccine this weekend. We should all do our part and get the shot to build herd immunity and curtail this horrible situation we all find ourselves in. Another bright note is my ability to be around my family more than I ever have since establishing my lab in New York away from my home in Connecticut. And that was 15 years ago when my son was still quite young! But I am still trying to answer the question. I have honed my skills in virtual speaking, is that considered a cool thing.



