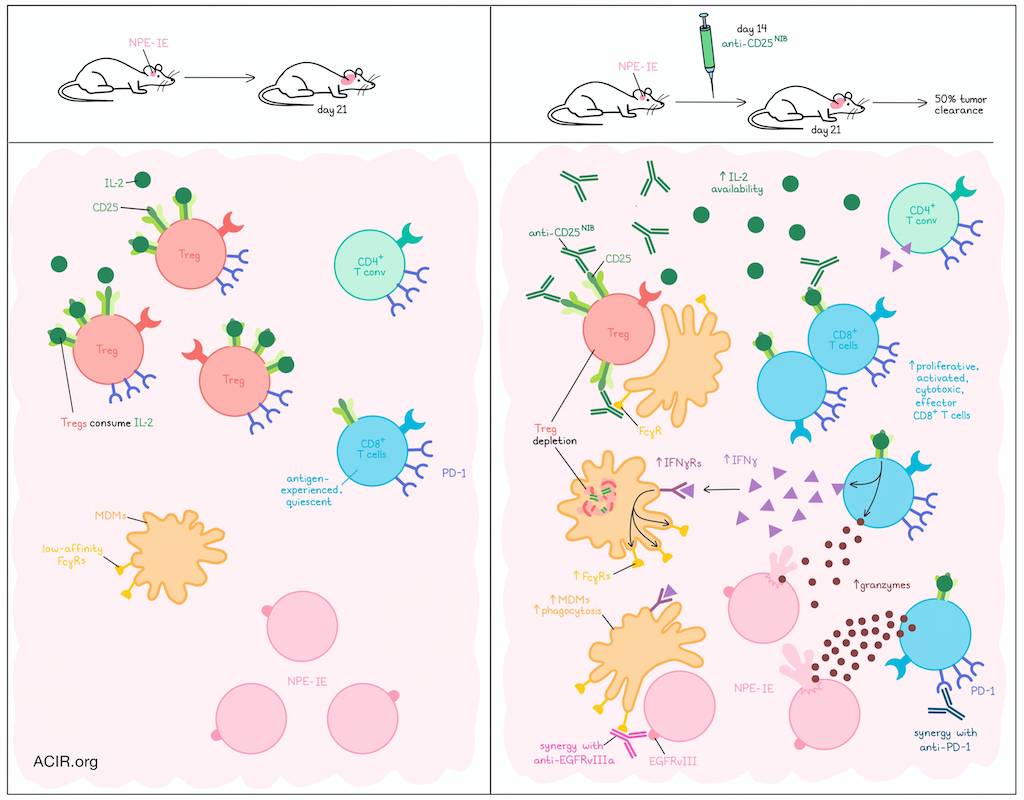
Glioblastoma and metastases in the brain are difficult to treat with immunotherapy, in part due to poor immune infiltration and a suppressive immune microenvironment, which can be marked by Tregs. Investigating ways of targeting Tregs, Galvez-Cancino et al. investigated using a non-interleukin-2 (IL-2) blocking (NIB) anti-CD25 antibodies (anti-CD25NIB) for Treg depletion in brain tumors. Their results were recently published in Immunity.
To first explore the role of Tregs in human glioblastoma, Galvez-Cancino et al. evaluated publicly available flow cytometry and scRNAseq datasets. Among lymphocytes, CD8+ T cells and conventional CD4+ T cells (CD4+ Tconv) expressing high levels of PD-1 were the most abundant, and appeared to be in antigen-experienced, but quiescent states, and were less proliferative than in other locations outside of the brain. Tregs were also abundant and were marked by high expression of CD25 and CTLA-4, though CD25 was found to be more Treg-specific.
Given that Tregs can consume IL-2, limiting its availability for CD8+ T cells and CD4+ Tconv, the researchers investigated antibody-mediated Treg depletion, which relies on FcγR engagement and phagocytosis by innate effector cells. To study this, the researchers evaluated an Nf1-/-Pten-/-EGFRvIII+ glioblastoma tumor line (NPE-IE) implanted into the brains of mice. After 3 weeks of growth, these tumors showed an abundance of CD8+ T cells, CD4+ Tconv cells, and Tregs, all expressing high levels of PD-1 and low levels of Ki67, mirroring the human datasets. When tumor-bearing mice were treated with anti-CD25NIB on day 14, Tregs and other lymphoid clusters were reduced on day 21, while proliferative, cytolytic, effector CD8+ T cells were increased. While this increased CD8+ T cell population did express CD25, it was at very low levels, suggesting that though they were still capable of sensing IL-2, they were less likely to be depleted. In line with this, neutralization of IL-2 prevented the increase in cytotoxic T cells, suggesting that it was mediated by the increased supply of IL-2 following Treg depletion. Importantly, anti-CD25NIB promoted tumor clearance in about 50% of mice, dependent on CD8+ T cells and IL-2.
Looking at T cell clonal expansion following anti-CD25NIB treatment, Galvez-Cancino et al. found that clones of cycling/proliferating CD8+ T cells were hyperexpanded, while clones of CD4+ Tconv cells were not. These CD8+ T cell clones largely accumulated in effector clusters and were marked by downregulation of Tox and upregulation of granzymes and proliferation-related genes. Increased TCR sharing was observed between cycling, effector, and progenitor exhausted clusters. Following evidence that anti-CD25NIB increased proliferating PD1+KI67hiGZMB+ cells, the researchers evaluated anti-CD25NIB in combination with anti-PD-1, and observed enhanced mouse survival.
Next, the researchers analyzed the innate immune cells involved in Treg depletion. Looking at FcγR expression, they found that monocyte-derived macrophages (MDMs) expressed the highest levels of low-affinity FcγRs, suggesting a strong potential to engage in antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). Indeed, FcγRhi MDMs were found to be critical to anti-CD25NIB-mediated Treg depletion, and increased in frequency following treatment, dependent on IL-2. As MDMs do not express the IL-2 receptor, the researchers hypothesized that increased access to IL-2 increased CD8+ T cell production of IFNγ. This was confirmed by results showing that expanding Mki67-expressing CD8+ T cells increased Ifng expression following treatment, and CD8+ T cells were required for the anti-CD25NIB-mediated increase in FcγRhi MDMs, with lesser contributions from CD4+ Tconv and NK cells. Spatial transcriptomics also showed significant enrichment of MDM, CD8+ T cell, CD4+ Tconv, and NK cell signatures in the proximity of Ifng transcripts.
Looking closer at FcγRhi MDMs following Treg depletion, the researchers observed markers indicative of IFN sensing and phagocytic potential in flow cytometry and RNAseq data for both mouse and human tumors. Experimentally, FcγRhi MDMs treated with anti-CD25NIB also showed increased evidence of phagocytosis when challenged in vitro with CFSE-loaded tumor cells. To capitalize on this increased phagocytic potential, the researchers tested anti-CD25NIB in combination with ADCC/ADCP-competent anti-EGFRvIII antibodies, and found that the combination further enhanced tumor regression and long-term survival in both brain and non-brain tumor models, dependent on IFNγ.
Finally, to evaluate the potential clinical relevance of their findings, Galvez-Cancino et al. evaluated human GBM samples, which had high frequencies of myeloid cells and lower frequencies of lymphoid cells. The patient samples showed some similarities with the mouse models in regards to FcγR expression on myeloid cells, though there was more variability in the patient samples. Still, CD49Dhi MDMs had the highest co-expression of FcγRIIa and FcγRIIb receptors and some of the highest levels of FcγRIII, IFNγR1 and IFNγR2. Upon stimulation of patient-samples with IFNγ, only CD49Dhi MDMs increased FcγRIIa, suggesting maturation and activation. Upon treatment of patient samples with anti-human CD25NIB, samples showed reduced fractions of CD25+ Tregs, which was associated with an increase in effector memory CD8+ T cells and granzyme B.
Together, these results suggested that Tregs play an immunosuppressive role in brain TMEs. Depleting Tregs with anti-CD25NIB increased and activated antitumor CD8+ T cells, dependent on an increased availability of IL-2. These CD8+ T cells showed increased production of IFNγ, which in turn, increased intratumoral MDMs and enhanced FcγR expression and phagocytosis. Based on these mechanisms, the researchers evaluated rational combinations with anti-PD-1 or anti-EGFRvIII antibodies, each of which synergized with anti-CD25NIB, improving tumor control and survival in mice. Analysis of human data suggested that these findings would likely translate to patients, and could be used to improve the treatment of brain malignancies in clinical settings.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Felipe Galvez-Cancino and lead author Sergio Quezada answered our questions.

What was the most surprising finding of this study for you?
FGC: The fact that Treg depletion can promote a recruitment of highly phagocytic myeloid cells is by far one of the most surprising and exciting findings of our research. These findings have the potential of informing novel therapeutic combinations aimed at harnessing the phagocytic capacity of these cells. As shown in the paper, we found that Treg depletion can enhance the efficacy of tumor-targeting antibodies that bind to surface antigens on tumor cells.
SQ: The activation of the myeloid compartment within brain tumours was an important and surprising finding. Linked to this is the fact that such activation offers a window of opportunity for the combination of Treg-depleting antibodies with tumor-targeting antibodies. This is a surprising new finding backed up by our mechanistic data. Most importantly, our data also suggest that these changes in the innate TME could be “wasted” if one would not act on them through the incorporation of tumor-targeting antibodies as a combo partner for Treg-depleting antibodies.
What is the outlook?
FGC: Treg depletion holds promise for developing novel therapies for GBM patients that deplete Tregs or target the IL-2 pathways, which are controlled by these cells within tumors. In addition, our work characterises the Fc gamma receptor landscape, identifying specific populations of macrophages as key mediators of antibody-dependent cellular phagocytosis, which has the potential for informing the development of novel immunotherapies.
SQ: The data also demonstrates the variability in Treg depletion in human brain tumors, and the need to better understand the innate TME, its role in the activity of depleting antibodies, and how to develop drugs and combinations to maximize innate activity (such as ADCC/ADCP) within brain tumors.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
FGC: This is a marathon, not a sprint; the experiments, paper writing, and revisions will take time. Just keep going until it is done.
What was the coolest thing you’ve learned (about) recently outside of work?
SQ: I just spent an amazing weekend with my son (boys weekend out) in Denmark learning about Lego at the Lego house in Bilund. I had no clue Lego was made of wood originally!



