CTLA-4 is a well established immune checkpoint with a key a role in limiting autoimmunity, but it exists in both membrane-bound (mCTLA-4) and soluble (sCTLA-4) forms, and the different sources, kinetics, and functions of sCTLA-4 in particular are not fully understood. In research recently published in Immunity, Osaki and Sakaguchi explored numerous aspects of sCTLA-4, identified Tregs as its primary source, and uncovered its role in suppressing type 1 immunity while allowing for maintenance of type 2 immunity in settings of chronic inflammation.
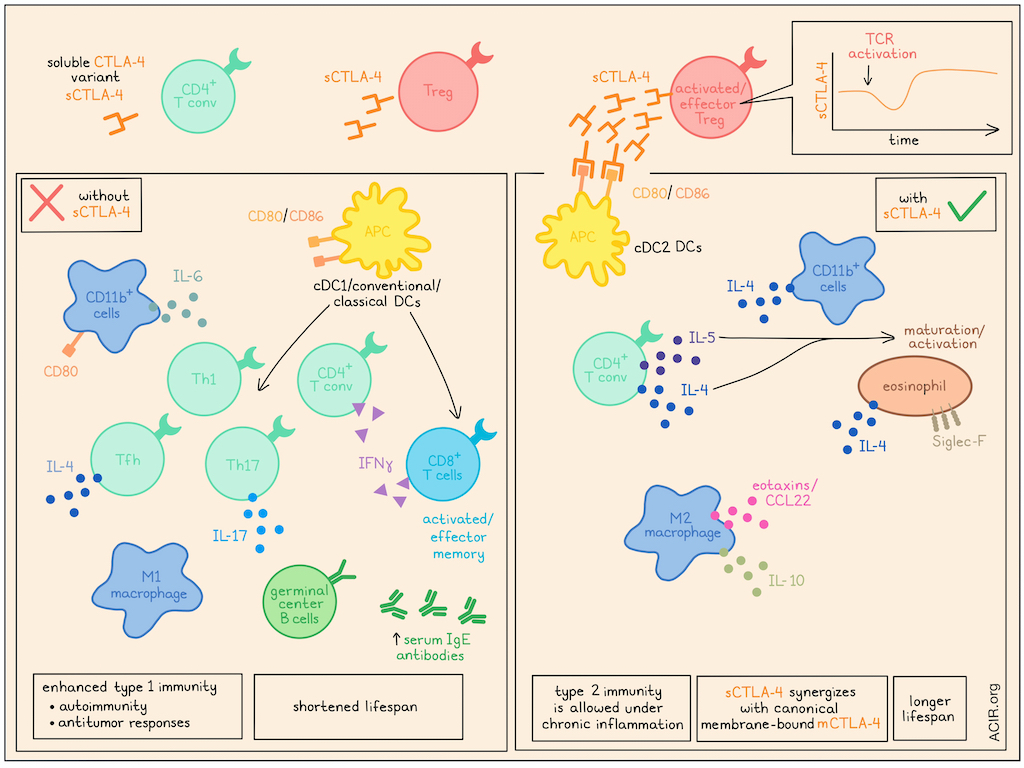
To begin, the researchers evaluated expression of sCTLA-4 in normal mice, and found that compared to conventional CD4+ T cells, Tregs showed increased expression of sCTLA-4, which was further increased in activated/effector Tregs, outside of lymphoid organs, and in chronic inflammatory settings, including colitis and tumors. TCR activation of Tregs initially reduced sCTLA-4 expression and instead increased mCTLA-4 expression, but over time, sCTLA-4 expression was restored and increased to above baseline, staying elevated for at least 3 weeks.
Investigating the role of sCTLA-4, the researchers generated a mouse model that was deficient in sCTLA-4, but maintained mCTLA-4 (S-M+). While these mice grew and survived normally over 30 weeks, they showed enlarged ectopic lymphoid structures in the adipose tissues, increased leukocyte infiltration into lungs at 30 weeks, and higher concentrations of serum antibodies (including IgEs), IL-6, and TNFα than wild-type S+M+ mice. There was also an increase in effector memory CD8+ and CD4+ T cells (including those secreting IFNγ), follicular helper T cells (including those secreting IL-4), IL-17+ CD4+ T cells, and germinal center B cells, and a decrease in Tregs. Macrophages expressed higher levels of M1 markers and lower levels of M2 markers. DCs were predominantly cDC1/classical conventional DCs, which are known to contribute to Th1 and Th17 induction. These results suggested that sCTLA-4 deficiency supported cDC1 differentiation to evoke type 1 immune responses, marked by Th1, Th17, Tfh, and Tc1 differentiation in CD4+ T cells, an M1-like phenotype in macrophages, increased antibody production, and a high potential for autoimmunity.
Next, the researchers generated a mouse model that expressed sCTLA-4 (at relatively high levels), but not mCTLA-4 (S+M-). While S-M- mice quickly developed fatal systemic immunity, S+M- mice were less debilitated and lived longer, but still showed signs of autoimmunity. While both S+M- and S-M- mice showed an increase in activated conventional CD4+ and CD8+ T cells relative to wild-type mice, Th1 and Th17 cells were lower in the mesenteric LNs of S+M- mice compared to S-M- mice. Further, S+M- mice showed increased concentrations in the serum of Th2 cytokines IL-4, IL-5, IL-10, M-CSF, but lower concentrations of IL-6 and IL-17A compared to S-M- mice. These results suggested that the presence of sCTLA-4 alleviated severe autoimmunity by reducing type 1 immunity, without disrupting type 2 immunity.
Osaki and Sakaguchi hypothesized that sCTLA-4 exerts immunosuppressive effects through its interactions with immune cells expressing CD80/CD86, such as CD11b+ cells (monocytes/macrophages/granulocytes), CD11c+ DCs, or B220+ B cells. Evaluating these immune populations in 3-week-old S+M- and S+M+ (wild-type) mice, the researchers found that while S-M- mice showed a notable increase in the proportion of CD80+CD11b+ cells among splenic CD11b+ cells (compared to S+M+ mice), there was no significant increase in S+M- mice. Cytokine analysis showed that splenic CD11b+ cells in S+M- mice expressed high levels of IL-4, while S-M- mice expressed high levels of IL-6. Further, macrophages from S+M- mice showed increased production of IL-10, expression of M2 markers, and genes related to tissue remodeling, and decreased genes related to the Th17 pathway compared to those from S-M- mice, which expressed higher levels of M1 markers.
Despite these phenotypic differences in macrophages from S+M- or S-M- mice, the expression levels of cytokine receptors were similar. Therefore, the researchers evaluated whether their differences could be attributed to differential exposure to Th2 cytokines. They found that treating macrophages with IL-4 and/or IL-10 increased CD206 and M2 marker expression and suppressed M1 marker expression, mimicking observations in S+M- mice. However, direct treatment of macrophages with sCTLA-4 did not have the same effect, suggesting that sCTLA-4 induces cytokine production by another immune cell type. They further found that in S+M- mice (compared to S-M- mice), DCs were more polarized towards cDC2s, and the frequencies of IL-4- and/or IL-5-producing circulating CD4+ T cells were increased, which appeared to drive the activation and maturation of eosinophils, increasing their expression of siglec-F and IL-4. Additionally, peritoneal macrophages expressed higher levels of eotaxins and CCL22, which were also increased in serum.
Investigating whether sCTLA-4 facilitates Th2 differentiation through interactions with CD80/CD86, the researchers evaluated the effects of a CTLA-4-Ig (positive control), sCTLA-4, and a mutant sCTLA-4 that lacks the binding domain for CD80/CD86 (Y139 sCTLA-4). In in vitro activation and differentiation assays, CTLA-4-Ig and sCTLA-4 both inhibited the proliferation of naive T cells and differentiation towards Th1, while Y139A sCTLA-4 did not. When IL-4 was added to induce Th2 conditions, sCTLA-4 allowed for Th2 differentiation, even at concentrations that suppressed Th1 differentiation. Similar results were observed with blockade of CD80, CD86, or both, with or without the addition of IL-4. These results suggest that sCTLA-4 blocks CD80/CD86 on APCs, limiting their capacity to activate T cells and induce Th1 differentiation, but not IL-4-induced Th2 differentiation.
Finally, Osaki and Sakaguchi examined the functions of sCTLA-4 in various murine disease models. They found that in an induced colitis model, both sCTLA-4 and mCTLA-4 protected mice from damage by inhibiting Th1/Th17 cell differentiation and expansion. Similarly, in an experimental asthma model, sCTLA-4 contributed to protection against more severe bronchoconstriction and peribronchiolar cellular infiltration, and in a wound healing model, sCTLA-4 contributed to proper wound healing. In murine cancer models. sCTLA-4 contributed to faster tumor growth and reduced infiltration of CD8+ T cells, particularly cytolytic CD8+ T cells, dependent on binding to CD80/CD86. It also contributed to lower IFNγ and a higher ratio of M2-like macrophages.
Overall, these results show that sCTLA-4 is produced predominantly by Tregs and plays a role in limiting autoimmunity though blocking CD80/86 on APCs, thus limiting the induction of type 1 immunity, while still allowing for the induction of type 2 immunity. However, in cancer settings, sCTLA-4 contributed to a less favorable TME and faster tumor growth, suggesting that it also limits antitumor immunity and could serve as a potential target, along with mCTLA-4, for cancer immunotherapy.
Write-up and image by Lauren Hitchings



