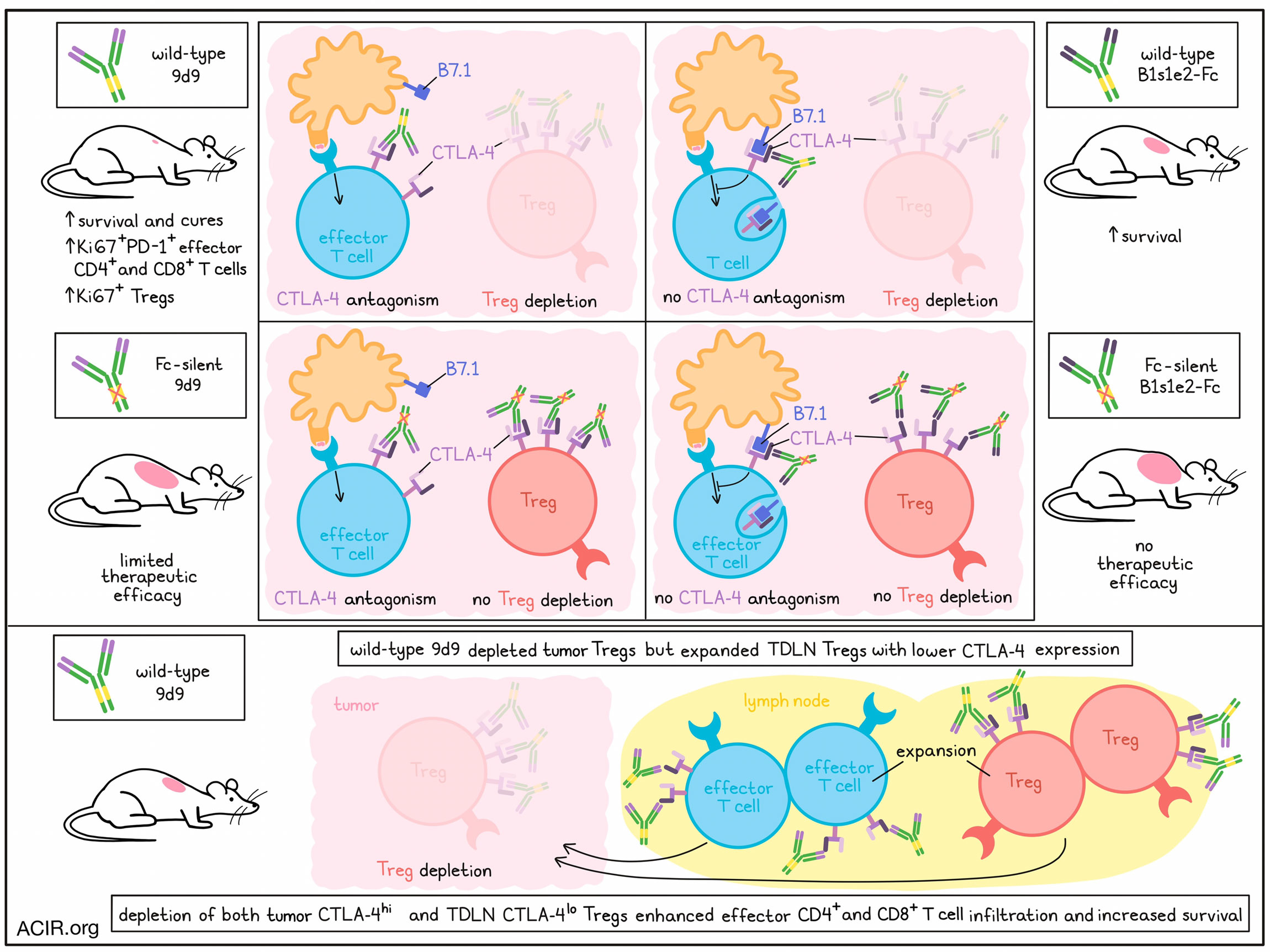
The mechanism of action of anti-CTLA-4 therapy remains incompletely understood. Hypotheses suggest that either antagonism of CTLA-4:B7 binding or Fc effector-mediated regulatory T cell (Treg) depletion might be responsible for the efficacy of the therapy. Lax et al. researched both these mechanisms with engineered antibodies, allowing testing of separate and combined antagonism and Treg depletion features to determine which of these mechanisms results in antitumor efficacy. Their results were recently published in PNAS.
The researchers used yeast surface display (YSD) with a library based on the DNA binding protein Sso7d as a scaffold to identify B7-noncompetitive CTLA-4 binders. Binders were selected and enriched for those with the highest affinity to CTLA-4-Fc; selection was done in the presence of excess B7.1-Fc to eliminate binders that targeted the B7.1 binding site on CTLA-4. This resulted in a single clone (b1s1), which had undetectable monomeric affinity to cell surface CTLA-4. After two rounds of affinity maturation, a dimeric mIgG2c-Fc fusion was prepared, creating the “non-antagonistic” b1s1e2-Fc. For comparison, the researchers used the “antagonistic” mIgG2c 9d9 antibody, which competitively inhibits the B7:CTLA-4 interaction. Both b1s1e2-Fc and 9d9 had similar affinity to cell surface CTLA-4, allowing comparisons of the therapies.
To confirm that b1s1e2-Fc bound to a distinct epitope from B7.1, a YSD epitope mapping system was developed with mutant libraries of CTLA-4 displayed on yeast surface, and CTLA-4 variants unable to bind either B7.1-Fc or b1s1e2-Fc were enriched. The b1s1e2-Fc epitope mapping suggested a single residue was required for binding, F67, which was present in a different loop of CTLA-4 than the canonical “MYPPPY” region known to bind B7, confirming a distinct epitope. When binding to CTLA-4WT and CTLA-4F67L was compared, 9d9 had equal binding to both, while b1s1e2-Fc only bound CTLA-4WT.
Since the extracellular domain of CTLA-4 is small, a distinct epitope may not be sufficient to be fully B7-noncompetitive if steric hindrance prevents binding of b1s1e2-Fc and B7. To test this, the researchers measured the reduction in binding affinity to CTLA-4 bound to a B7.1-coated plate and compared it to a CTLA-4-coated plate. This showed that b1s1e2-Fc could bind CTLA-4, even when CTLA-4 was bound to B7.1, while binding of 9d9 was reduced in that situation. As CTLA-4 antagonism blocks the CTLA-4:B7 axis, as well as CTLA-4 from transendocytosing B7, the binding of b1s1e2-Fc on B7 transendocytosis was measured by incubating CTLA-4-expressing recipient cells with donor cells with either B7.1 or B7.2 in the presence of CTLA-4 binders. Remaining ligand after overnight incubation was similar for b1s1e2-Fc or no antibody, while 9d9 resulted in increased amounts of B7.1 and B7.2, suggesting b1s1e2-Fc does not functionally block CTLA-4 or inhibit transendocytosis.
Moving to the assessment of Treg depletion, the researchers made use of the MC38 colon carcinoma mouse model. The mice were subcutaneously (SC) inoculated with tumor cells, after which they received doses of the constructs intraperitoneally (IP) or intratumorally (IT) on day 7. One day later, the tumors were analyzed. Both b1s1e2-Fc and 9d9 depleted Tregs in the tumor. The researchers also created silent Fc versions of both constructs by introducing LALA-PG mutations into the Fc, to remove binding to FcγRs. These formats of the constructs did not deplete Tregs.
Next, the therapeutic efficacy of each construct and their silent Fc formats were determined. Mice with SC MC38 tumors were dosed IT or IP with the constructs on days 7, 10, and 14, and survival was assessed. Mice that were treated IT with 9d9 WT or b1s1e2-Fc WT had a significantly improved survival compared to control mice, but 9d9 WT led to more cures (47%) than b1s1e2-Fc (0%). The 9d9 silenced Fc resulted in similar survival as control mice, suggesting CTLA-4 antagonism alone is not an effective therapy. As b1s1e2-Fc does not antagonize CTLA-4, silencing Fc completely removed the therapeutic efficacy. IP dosing had similar effects as IT treatment. Overall, there was greater benefit from 9d9 WT than that of b1s1e2-Fc, suggesting both CTLA-4 antagonism and intratumoral Treg depletion results in the best responses.
Next, the researchers assessed the effects of treatment on tumor-draining lymph nodes (TDLN). Mice bearing MC38 were dosed on day 7 and TDLN were assessed on day 10. Mice treated with 9d9 had increased frequencies of Ki67+PD-1+ CD4+ and CD8+ effector T cells and Ki67+ Tregs compared to control or b1s1e2-Fc treatment, suggesting CTLA-4 antagonism is required for enhanced T cell priming.
In control mice, the percentage of CTLA-4+ Tregs and the level of CTLA-4 expression were higher for Tregs in the tumor than those in the TDLN. Since the TDLN Tregs do not have sufficient levels of CTLA-4 on the surface to induce depletion, 9d9 treatment results in priming of these Tregs. Treatment with 9d9 resulted in Treg expansion to a greater degree than effector T cells at 3 days post-treatment in the TDLN, which may be due to higher CTLA-4 expression levels on Tregs than effector T cells. As this may inhibit some of the priming of effector T cells, the researchers hypothesized that depleting TDLN Tregs, in addition to tumoral Tregs, might improve efficacy. The researchers made use of the Foxp3-DTR mouse model to assess this. This model allows for selective Treg depletion via the administration of diphtheria toxin (DT). Using a low-dose IT DT dosing for local Treg depletion, it was shown that local Treg depletion led to an increase in intratumoral effector CD4+ T cells, as well as a non-significant increase in effector CD8+ T cells. There were no differences in the frequency of CD4+ or CD8+ effector T cells in the TDLN. Using this model, mice inoculated with MC38 or B16F10 tumors and treated with DT starting at day 7, dosed every two days to maintain Treg depletion in tumors and TDLNs, had improved survival compared to control mice.
The data presented in this study suggest that both Treg depletion and CTLA-4 antagonism are vital for the efficacy of this treatment. Treg depletion in both the tumor and TDLN can result in more effective priming of T cells; however, current clinical antibodies targeting CTLA-4 do not target TDLN Tregs due to their low CTLA-4 expression. Therefore, the results of this study imply that novel therapies that induce depletion of these Tregs might be a good combination strategy to help increase the efficacy of anti-CTLA-4 treatment.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, first author Brianna M. Lax and lead author K. Dane Wittrup answered our questions.

What was the most surprising finding of this study for you?
BML: One of the most surprising findings to me was that intratumoral and nodal Treg depletion using diphtheria toxin was more efficacious than traditional anti-CTLA-4 antibodies. Although we showed that T cell priming in the lymph node is critical for efficacy, I wasn’t expecting the associated nodal Treg proliferation to be so detrimental. The diphtheria toxin studies showed just how powerful Tregs in the lymph node are at suppressing the immune system.
KDW: I was surprised at how ineffective depletion of Tregs in the lymph node is with anti-CTLA-4 antibodies, most likely due to the low steady-state surface levels of CTLA-4 in that compartment by contrast to the tumor. This is problematic, given the significance of lymph node-resident Tregs in suppressing CD8+ T cell priming.
What is the outlook?
BML: These findings provide valuable insights for the next generation of checkpoint inhibitors. This work supports the development of human anti-CTLA-4 antibodies with enhanced Treg depletion capabilities while also highlighting the therapeutic potential of depleting nodal Tregs and providing motivation for the development of therapies that can do so.
KDW: In the end, Bri’s very cool noncompetitive CTLA-4 binder didn’t crack the nut of lymph node Treg depletion. However, I believe it did shed light on the complex requirement for both CTLA-4 ligand antagonism and Treg depletion for therapeutic efficacy, and as such, could help future antibody developers craft alternative strategies.
What was the coolest thing you’ve learned (about) recently outside of work?
BML: I recently traveled to Oaxaca, Mexico and swam in the bioluminescent lagoon in Chacahua National Park, where I learned that these magical plankton can only live in brackish water that is 70% saltwater and 30% freshwater. These organisms use luciferases to catalyze oxygenation reactions that release light with a shocking efficiency of around 98%! These creatures are proving that enzymes are the coolest type of protein out there.
KDW: Four months of wearing a Fitbit have shown me that I’m not as self-disciplined as I’d thought I was. Without altering my putative workout routine, just letting this tyrannical watch shame me into full compliance has produced improved blood pressure and resting heart rate. (These are the things a 60-year old professor gets excited about instead of swimming with magical plankton, I guess.)



