Predicting whether a patient will respond to immune checkpoint blockade (ICB) and identifying resistance regulators are key research goals in cancer immunotherapy. With current biomarkers still falling short of accurate prediction, Jiang, Gu, and Pan et al. developed a computational method called Tumor Immune Dysfunction and Exclusion (TIDE) that models mechanisms of tumor immune escape based on treatment-naive data from over 33,000 samples taken across 189 studies, and predicts response to ICB.
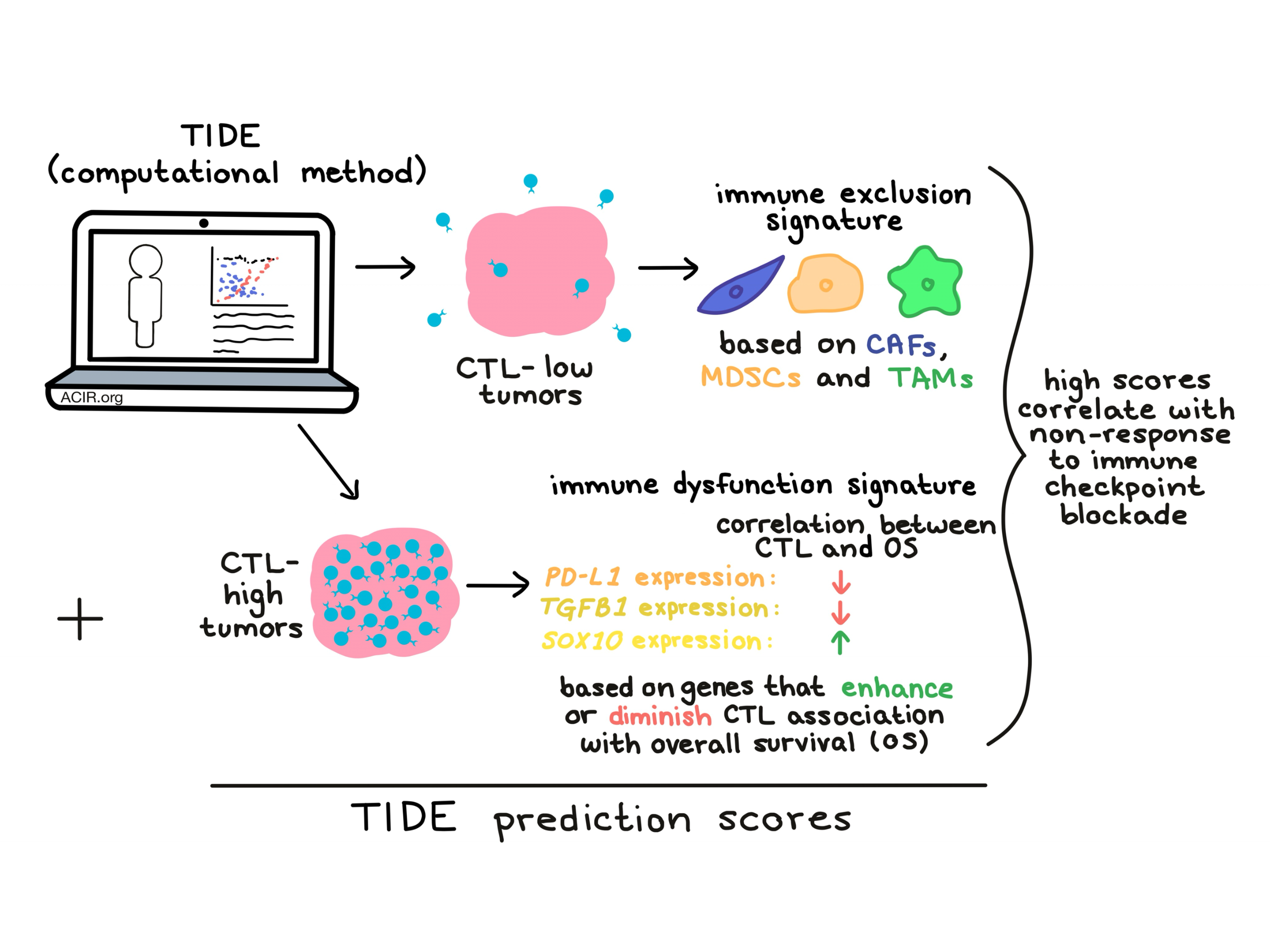
Tumors typically escape the immune system by either preventing cytotoxic T lymphocyte (CTL) infiltration, or by inducing dysfunction in CTLs that do manage to infiltrate, and TIDE was developed as a way to identify gene signatures of both T cell dysfunction and exclusion. To identify gene signatures of dysfunction, the researchers used the average expression level of CD8A, CD8B, GZMA, GZMB, and PRF1 to estimate the level of CTL infiltration in a tumor, and then selected a cutoff point (initially the average of all samples) at which to identify low- and high-CTL levels in tumors. CTL level correlated with overall survival.
The researchers then went on to use statistical methods to identify genes in CTL-high tumors that “interacted” with CTL level, meaning that if a particular gene was upregulated, it either strengthened or weakened the association between CTL level and overall survival. Genes like TGFB1 and PD-L1 were found to negatively interact with CTL levels, while genes like SOX10 positively interacted with CTL level. Notably, the method is agnostic to the cell source of the expressed gene. Together this data could be used to identify T cell dysfunction signatures for individual tumors, and T cell dysfunction scores for different cancer data sets. High TIDE T cell dysfunction scores were consistent with late-stage T cell dysfunction (a T cell state identified in prior studies as unable to be rescued by ICB) and signatures of immune evasion, with upregulation of pathways related to inflammation and response to interferons, and downregulated pathways related to mTORC1 signaling, protein secretion, and glycolysis. Averaging TIDE dysfunction scores from different cohorts gave the best performance.
In CTL-low tumors, the researchers searched for signatures of T cell exclusion from the tumor microenvironment. They looked at transcriptomic signatures of three exclusionary cell types: cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), and M2 tumor-associated macrophages (TAMs) and derived signatures of T cell exclusion using expression profiles of these cell types. Higher immune exclusion scores correlated negatively with CTL levels across all solid tumor data sets.
Not unexpectedly, T cell dysfunction scores correlated inversely with T cell exclusion scores, and the researchers hypothesized that integrating the two signatures might predict whether a patient would respond to ICB. For these studies they focused on melanoma, as this was the only tumor type that had available data on both tumor expression and clinical outcomes for patients treated with anti-PD-1 and anti-CTLA-4. In patients with CTL-high tumors, those whose tumor expression data correlated strongly with T cell dysfunction could be predicted as non-responders. Similarly, in patients with CTL-low tumors, tumor expression data that correlated strongly with the T cell exclusion signature predicted non-response to ICB. These relationships were integrated to generate TIDE prediction scores (a single measure reflecting multiple variables), which allowed the researchers to rank tumors by their likeliness to respond to ICB.
Compared to widely used ICB biomarkers, including tumor mutation burden and PD-L1 expression, TIDE prediction scores were consistently better at predicting the efficacy of both anti-PD-1 and anti-CTLA-4, using either RNA-Seq or NanoString data. Further, TIDE prediction scores were able to predict overall survival under treatment, demonstrating the potential of TIDE as a prognostic tool. The researchers speculate that TIDE prediction scores may outperform other biomarkers because they account for both T cell dysfunction and T cell exclusion, while most other biomarkers address only one of these factors.
In addition to predicting response to immunotherapy, Jiang, Gu, and Pan et al. used TIDE to search for novel resistance regulators. Looking for genes that had high T cell dysfunction scores and were upregulated in an independent set of transcriptomic data from mice that developed resistance to anti-CTLA-4, the researchers identified SERPINB9 – a gene whose encoded protein can inactivate granzyme B – as the most upregulated gene, also noting that it was typically expressed at higher levels in non-responders. SERPINB9 expression was found to be upregulated in many cancers and independently associated with worse overall survival in two clinical studies of anti-CTLA-4. In vitro studies of knockout or overexpression of SERPINB9 confirmed the gene’s role in conferring resistance to T cell-mediated killing. They also showed that SERPINB9 expression could be induced following exposure to IFNγ. Together these results suggest that SERPINB9 could be expressed as a defensive mechanism in response to T cell attack and that it regulates resistance to ICB.
As a computational tool, TIDE offers unique insights into tumor immune evasion as well as response and resistance to checkpoint blockade. Because TIDE is trained using immunotherapy treatment-naive tumor samples, its application may be relevant in predicting response to immunotherapy even in cancer types that have little clinical data available on checkpoint blockade. To make TIDE widely and immediately available to clinicians and the public, the researchers have created a web application that generates a response prediction from an uploaded gene expression profile.
by Lauren Hitchings



