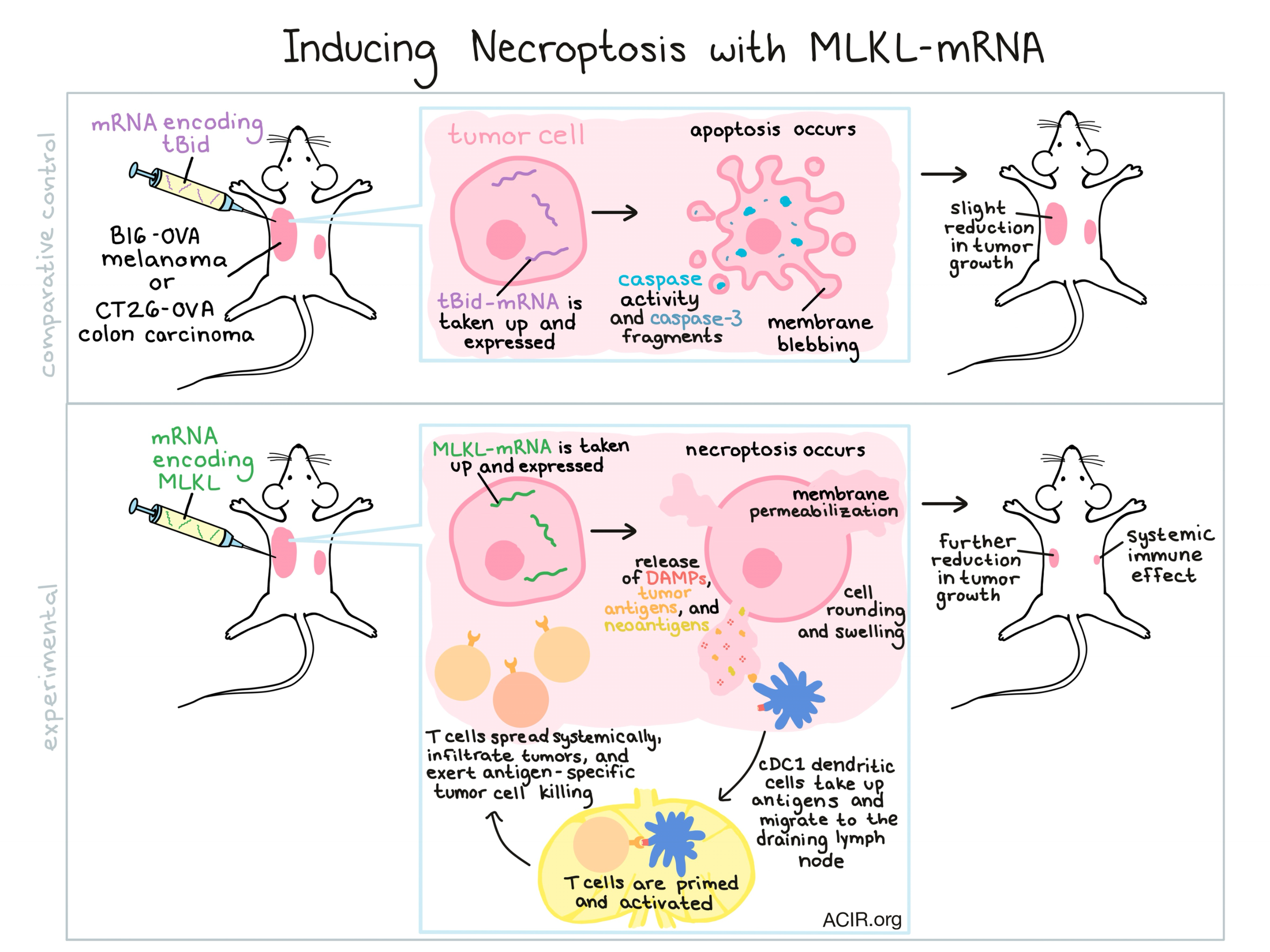
The cancer immunity cycle is the body’s natural way of staving off cancer, but in instances when cancer has already escaped this cycle, inducing immunogenic tumor cell death may be an effective way to reboot the system. In a recent study published in Nature Communications, Van Hoecke et al. explored the use of intratumorally delivered mRNA encoding the necroptotic cell death executioner protein MLKL (MLKL-mRNA) to induce necroptosis and a subsequent antitumor immune response.
To begin, Van Hoecke et al. generated and delivered mRNA encoding MLKL, tBid (an inducer of apoptosis), or Fluc (an irrelevant reporter) to B16 melanoma cells. The MLKL-mRNA induced necroptosis (marked by a lack of caspase activity or caspase-3 processing, followed by cell rounding, swelling, and membrane permeabilization), while tBid induced apoptosis (marked by blebbing, caspase activity, cleavage fragments of caspase-3, and loss of membrane integrity). MLKL-mRNA and tBid-mRNA both induced similar levels of tumor cell death, which was a three-fold increase over the delivery of control mRNA or saline.
In mice bearing B16 melanoma, intratumorally delivered mRNA was taken up by tumor cells, with peak protein expression occurring 12 hours after electroporation. In both B16-OVA melanoma and CT26-OVA colon carcinoma models, tBid-mRNA delayed tumor growth and increased median survival. MLKL-mRNA delayed tumor growth more significantly and improved survival further. MLKL-mRNA also outperformed repeated treatments of doxorubicin, a known apoptosis inducer, with fewer negative side effects.
Since in vitro results had shown that tBid-mRNA and MLKL-mRNA induced similar levels of tumor cell death, the researchers suspected that the difference in antitumor efficacy might be due to participation by the immune system following the release of damage-associated molecular patterns (DAMPs) and tumor antigens. To test whether there was a systemic immune effect occurring, the researchers treated tumor-bearing mice, then removed the primary tumor and rechallenged mice in the same location. They found that treatment with MLKL-mRNA protected 40% of B16 melanoma-bearing mice and 100% of CT26 colon carcinoma-bearing mice. Similarly, the researchers tested for an abscopal effect by implanting a primary tumor on one side, and later, a secondary tumor on the opposite side. Treatment of the primary tumor with MLKL-mRNA significantly delayed the growth of both primary and secondary tumors, indicating a systemic immune effect; this effect was weak in tBid-mRNA-treated mice and not observed in controls.
To further investigate the immune response involved in the antitumor efficacy of MLKL-mRNA, the researchers moved to determine whether the MLKL-induced necroptosis primes T cell responses. To this end, the researchers adoptively transferred reporter-labeled, OVA-specific CD4+ and CD8+ T cells into mice bearing B16-OVA melanoma. When they removed the lymph nodes of MLKL-mRNA-treated animals, they found evidence of T cell proliferation and increased numbers of OVA-specific CD4+ and CD8+ T cells. An in vivo killing assay showed that MLKL-mRNA increased specific killing to 75% of target cells, while tBid-mRNA induced only 40% target cell killing, and controls induced no significant target cell lysis. Further, the researchers were able to identify T cells from MLKL-mRNA-treated mice that were specific to known neoepitopes in both B16 melanoma and CT26 colon carcinoma, but not the wild-type proteins. These neoepitope-specific responses were not identified in mice treated with tBid-mRNA or controls.
With evidence for the participation of T cells in the antitumor efficacy of MLKL-mRNA, the researchers next explored the mechanism by which the T cells become activated. Following MLKL-mRNA treatment, the researchers observed an influx of cDC1 (Batf3-dependent) and cDC2 (IRF4-dependent) dendritic cells into the tumor bed and tumor draining lymph nodes. Various knockout models revealed that the presence of cDC1 dendritic cells, type I interferon signaling, and migration of dendritic cells to the draining lymph node were were all required in order to mount T cell priming and a subsequent OVA-specific cytotoxic immune response.
Because intratumoral treatment with MLKL-mRNA increases T cell priming and infiltration into the tumor, Van Hoeke et al. hypothesized that their treatment would synergize with anti-PD-1 blockade, which would overcome T cell suppression in the tumor. They tested their hypothesis in mice with primary and abscopal tumors. Treatment of the primary tumor with MLKL-mRNA combined with anti-PD-1 treatment led to significantly improved tumor suppression in both the primary tumor and the distant untreated tumor, and improved survival compared to either monotherapy.
To get a better idea of whether this therapy could translate to the clinic, the researchers assessed mRNA encoding for human MLKL and found that it could effectively kill human cancer cells in vitro. In a humanized mouse model bearing human follicular lymphoma tumors, hMLKL-mRNA induced a strong antitumor effect that significantly delayed tumor growth and increased median survival, outperforming tBid-mRNA treatment and controls. While the preclinical data is promising, further preclinical research is necessary to fully understand the mechanism of MLKL-induced necroptosis. Clinical translation would also come with a host of additional hurdles, including the accessibility of tumors and their larger size. Still, evidence that MLKL-encoding mRNA can act as an in situ vaccine presents an exciting new avenue for immunotherapy research.
by Lauren Hitchings



