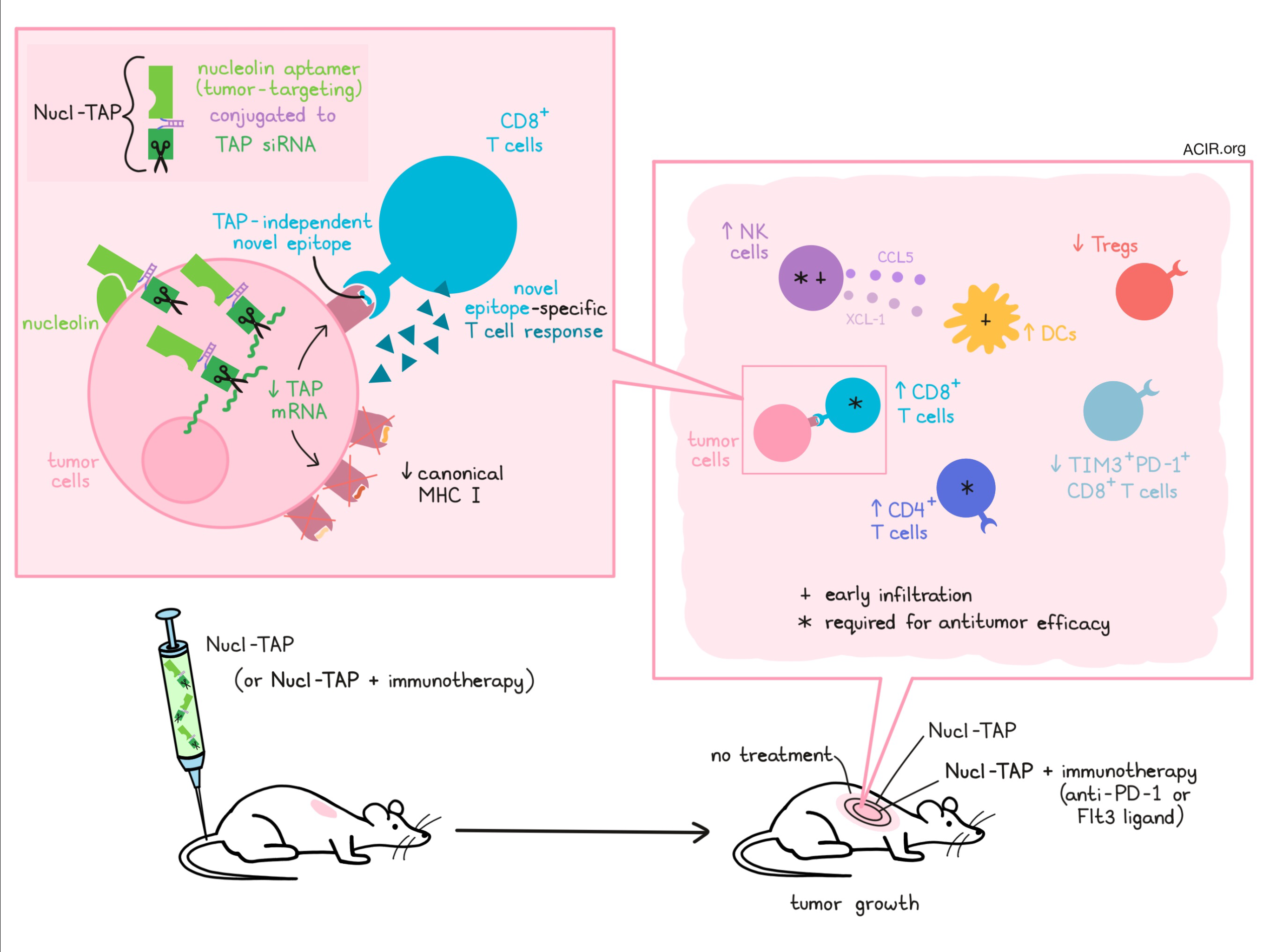
Tumors with high neoantigen burdens tend to respond well to immunotherapy, but patients whose tumors express few or no neoantigens may not derive the same benefits. Enhancing the immunogenicity of tumors in a way that is clinically relevant and broadly translatable could be a way to enhance immunotherapy response rates. Building on previous research, which showed that tumor cells deficient in TAP (the peptide transporter associated with antigen processing) express non-mutation-based neoantigens from common housekeeping genes, Garrido et al. set out to knock down TAP in situ to enhance tumor immunogenicity and induce antitumor immunity. Their results were reported in Nature Communications.
In an effort to specifically silence TAP in tumors, Garrido et al. conjugated a TAP-targeting siRNA to an oligonucleotide aptamer targeting nucleolin – a protein found on the surface of many tumor cells of mouse and human origin. In vitro, this Nucl-TAP conjugate induced downregulation of TAP mRNA and downregulation of canonical MHC class I alleles (but not the oligomorphic HLA-E/Qa-1 allele) in several tumor cell lines, recapitulating previous observations of TAP-deficient tumor cells. Nucl-TAP-treated cells also induced activation of T cells reactive to the TRH4 epitope (an epitope previously observed to arise on the downregulated canonical MHC in TAP-deficient cells), suggesting that Nucl-TAP induced presentation of this neoepitope on tumor cells.
Exploring the effects of Nucl-TAP in vivo, the researchers administered the conjugate molecule in a number of tumor models. They found that Nucl-TAP accumulated preferentially in subcutaneous tumors (observed in 4T1 tumor model) and reduced canonical MHC-Ia expression in tumor cells (observed in A20 lymphoma model). It also reduced the growth of palpable 4T1, RMA T lymphoma, and A20 tumors, and even induced complete rejection of some A20 tumors. In the 4T1 tumor model, the addition of anti-PD-1 or Flt3 ligand enhanced the antitumor efficacy. In a KPC-derived pancreatic tumor model, Nucl-TAP reduced local growth and metastasis, and was comparable to the leading therapeutic agent for that model, Minnelide. Nucl-TAP also sensitized tamoxifen-induced BRAF/PTEN melanoma to anti-PD-1 or Flt3 ligand immunotherapy. In an MC38 colon carcinoma model, treatment with Nucl-TAP induced antitumor effects that were comparable to antitumor effects induced by a vaccine targeting three prototypic mutation-based neoantigens.
To show that the tumor control induced by Nucl-TAP was in fact mediated by an immune response, Garrido et al. performed a series of antibody depletion experiments showing that in 4T1-bearing mice, the antitumor effect of Nucl-TAP was dependent on NK cells, CD4+ T cells, and CD8+ T cells. Consistent with a role for NK cells in recruiting DCs and indirectly promoting T cell responses, the researchers found that tumor infiltration by NK cells and DCs preceded infiltration by CD8+ T cells, and that tumors secreted the NK cell-derived DC chemoattractants XCL-1 and CCL5 in the early tumor microenvironment. Treatment with Nucl-TAP also increased CD4+ T cells, reduced Tregs, reduced TIM3+PD-1+ exhausted CD8+ T cells, and increased the CD8/Treg ratio in the tumor microenvironment of 4T1 tumors, pointing to a broad remodeling of the TME.
To determine whether the antitumor immune response was mediated by T cells specific for common TAP-deficiency-induced neoantigens, the researchers used tetramers and IFNγ induction to show that T cells from MC38 tumor-bearing mice treated with Nucl-TAP were reactive to the MHC-I-restricted TRH4 epitope and the Qa-1b-restricted FAP epitope, and were cytotoxic in vivo against splenocytes pulsed with the TRH4 peptide. As evidence that this antigen is commonly expressed across Nucl-TAP-treated tumors, the researchers showed that CD8+ T cells isolated from Nucl-TAP-treated MC38 tumors were also reactive against Nucl-TAP-treated RMA tumors. Further, Nucl-TAP treatment was shown to induce epitope spreading, as CD8+ T cells isolated from Nucl-TAP-treated RMA also had an antitumor effect when adoptively transferred into untreated mice bearing RMA tumors. Similar evidence of epitope spreading was observed in a 67NR breast carcinoma model.
No evidence of toxicity or off-target inflammation was observed in 4T1 tumor-bearing mice treated as many as six times with Nucl-TAP, nor in mice treated three times with Nucl-TAP plus anti-PD-1 or Flt3 ligand. In fact, Nucl-TAP appeared to demonstrate a safer profile than anti-CTLA-4. Whether autoimmunity may develop with long-term use, however, remains to be studied.
To explore whether this method might translate to humans, Garrido et al. developed an siRNA for human TAP and generated a human Nucl-TAP conjugate. In human tumor cells of melanoma and renal cell origins, Nucl-TAP downregulated TAP mRNA and HLA-expression. Based on prior evidence that human TAP-deficient tumor cells present a novel HLA-A2-restricted peptide derived from the LRPAP1 protein, the researchers found that Nucl-TAP-treated tumor cells induced activation of T cells specific to this peptide. This indicates that Nucl-TAP silences TAP and induces presentation of a common neoantigen in human tumor cells.
Overall, Garrido et al. show in mice and in human cells that a nucleolin aptamer conjugated to TAP siRNA can be broadly applied to specifically induce predictable neoantigens in tumor cells, thereby increasing the antigenicity of tumor lesions and enhancing the efficacy of immunotherapies. This strategy could be implemented to improve responses to immunotherapy or broaden the pool of potential responders.
by Lauren Hitchings
Meet the Researcher
This week, first author Greta Garrido and lead author Eli Gilboa answered our questions.

What prompted you to tackle this research question?
Greta & Eli: We have been interested in developing cancer vaccines, and recent experiences with checkpoint blockades have underscored the importance of targeting antigens unique to the tumor cells – neoantigens – that have not induced tolerance and and are not likely to elicit autoimmune pathologies. The main challenges that have been encountered in targeting neoantigens include that tumor cell-expressed neoantigens differ from patient to patient; that isolating neoantigens requires laborious, patient-specific, yet-to-be-perfected protocols; and that only a proportion of cancer patients express such neoantigens to begin with. With this in mind, we developed an approach to “decorate” tumor cells in situ with experimentally induced new antigens, in effect to make the tumor cells more “visible” to the immune system. Importantly, to do that in way that would be simple, inexpensive, and broadly applicable to virtually every cancer patient, we used a short chemically-synthesized oligonucleotide administered by injection.
What was the most surprising finding of this study for you?
Greta & Eli: How well it worked in multiple disparate murine tumor models. Just decorating tumor cells with new antigens was as effective as vaccinating against existing “classical” neoantigens. The applicability of this approach is dependent on the efficiency of delivering the therapeutic cargo to a large proportion of disseminated tumor cells in the body. It was (almost) too good to be true that in that one particular experimental system, tested efficiency approached 100%.
What was the coolest thing you’ve learned (about) recently outside of work?
Greta: Outside of the lab, my most exciting “discoveries” always come from my son. He is 5 years old, at this magic age when they have a lot of questions, but also incredible ideas. I have noticed that kid’s imaginations don’t have limits. They are building in their minds our short-term future. The other day he told me, “Mom, I want to remember you forever. So let me take a picture of you with my brain”. He used his eyes like a camera and he said, “It’s fine. Let me send it to you”. For that, he closed his eyes and asked me to focus. Finally, he asked me to check out if I had received it. Sure, I said, and took a picture of him with my eyes too. That happened at the same time I was reading Origin by Dan Brown and his findings concerning our destiny as a human race. He suggests artificial intelligence could become avatar for real people. So maybe it will be sooner that we expect.



